 |
Journal of Regional Section of Serbian Medical Association in Zajecar
Year 2006
Volumen 31 No 4 |
|
|
|
|
[ Home ] [ Gore/Up ][ <<< ] [ >>> ]
|
|
|
UDK: 616.1-056.7-053.6 |
ISSN 0350-2899 31 (2006) 4 p.
176-179 |
|
| |
Case reportPuberty Onset Congenital Erythropoietic Porphyria - A
Case Report
Sabhiya Majid (1), Qazi Massod Ahmad (2), Iffat Hassan (2),
Syed Nissar (3), Farah Sameena (2), Massarat Rasool (1)
(1) Department of Beiochemistry Government College and Associated
Hostpital Srinagar, Jammu and Kashmir, India, (2) Department of
Dermatology Government Medical College and Associated Hostpital
Srinagar, Jammu and Kashmir, India, (3) Department of Endocrinology S.
K. Institute of Medical Sciences Soura, Jammu and Kashmir, India |
|
| |
|
|
| |
Summary:
Congenital Erythropoietic Porphyria (CEP) or Gunther’s disease is an
extremely rare, autosomal, recessive, inheritable disorder of heme
metabolism. Primary abnormality in CEP is decreased uroporphyrinogen III
cosynthase activity resulting in accumulation and hyperexcretion of
biologically inactive type I porphyrins. Characteristic pink-red
fluorescence is observed in teeth, urine, feces, plasma and erythrocytes
when exposed to long wave ultra violet light under Wood’s lamp.
Clinically it is a non-acute type of porphyria, defect is expressed in
infancy and characteristic clinical features include extreme cutaneous
photosensitivity, blistering, scarring, milia formation, hypo- and hyper
pigmentation of photo exposed parts. Haemolytic anaemia with
splenomegaly, acro-osteolysis and retarded growth may be present. Life
span is usually decreased [1-7]. Only approximately 200 cases of CEP
have been reported till recent past globally. About 12 cases of adult
onset porphyria have been reported till date [8-15]. Here we are
reporting on the CEP patient with haemolytic anaemia. In this Kashmiri
boy the disease onset was around puberty.
Key words: Congenital Erythropoietic Porphyria, Porphyria
Profile, Photosensitivity.
Napomena: sažetak na srpskom jeziku
Note: summary in Serbian
|
|
| |
CASE REPORT
16 year old male, born of consanguineus marriage as full term normal
delivery, with normal milestone and development presented with the
history of skin lesions on exposed parts of body for the past 2 years
and passage of red colored urine for the past 6 months only. Striking
features on physical examination were a pinched nose, palor and brownish
discoloration of teeth which fluoresced intensely under UV-A light,
there was onycholysis of the right little finger. Systemic examination
revealed splenomegaly, rest of systemic examination was non
contributory. On cutaneous examination hypertrichosis was observed all
over the face excepting the periorbital areas. Skin lesions in the form
of vesicles, bullae were observed on exposed parts of face, hands and
feet. Healing of the lesions occurred with scarring, hyper and
hypopigmentation. Milia were present over knuckles with pigmentation in
the same area. These symptoms were suggestive of porphyria. Many
symptoms of porphyria are very similar to those experienced in other
more common diseases, thus laboratory testing based on the definite
pattern of accumulation and hyperexcretion of Porphyrins and porphyrin
precursor is most effective for diagnosis and typing of porphyrias. This
pattern of excretion in turn depends on the defective enzyme of heme
biosynthetic pathway and its site of expression. [3,4] Exact porphyrin
isomer identification needs sophisticated equipment like HPLC [10,11,15]
unavailable in our setup as it is in most hospitals in developing
countries. Hence, type of porphyrins increased and not exact isomer was
considered. Their preliminary extraction and differentiation by solvent
partition was followed by spectrophotometric measurement [3,5,10-16].
Thus investigations included USG abdomen , Chest X-ray, routine
hematological and biochemical tests, and the ‘Porphyria profile’
comprising of qualitative and quantitative analysis of porphyrins in
urine (24h), stool, plasma and erythrocytes and porphyrin precursors
(delta-aminolevaleunic acid and porphobilinogen) in urine.
Routine laboratory investigations significantly revealed decreased Hb
(8.0 g/dl), increased reticulocyte count (7% of circulating
erythrocytes). In peripheral blood film erythrocytes exhibited
anisocytosis, a dimorphic picture (microcytic and normrcytic type ),
hypochromia with poikilocytosis. Coombs test was negative. Platelets and
WBC were normal. LDH was increased (514 U/l). S. Iron was increased
(180ug /dl) with normal total iron binding capacity (350 ug/dl). Liver
function tests showed an elevated serum bilirubin with raised alkaline
phosphatase levels. All other routine investigations were within normal
limits.
As per porphyria profile Porphobilinogen and delta amino levaleunate was
absent in urine. Porphyrin levels were raised in urine, stool and blood
as observed by bright red fluorescence under Wood’s lamp. Quantitive
studies revealed that urinary porphyrins were elevated 50 times than
normal (uroporphyrin levels being more than coproporphyrins). Fecal
porphyrin levels too were greatly elevated (coproporphyrin levels were
higher than uroporphyrins). Erythrocytes showed greatly increased levels
of coproporphyrins with much lesser amounts of uroporphyrins,
protoporphyrins were negligible. Firm diagnosis was established on the
basis of this pattern of increase in porphyrin levels and clinical
picture by a systematic ruling out mechanism. The patient was not on any
prior medication. |
|
| |
|
|
| |
DISCUSSIONS
Congenital Erythropoietic Porphyria (CEP) or Gunther’s disease is an
extremely rare, autosomal, recessive, inheritable disorder of heme
metabolism. Clinically a non-acute type of porphyria, defect expressed
in infancy and is exacerbated by exposure to sunlight. Primary
abnormality in CEP is decreased uroporphyrinogen III cosynthase activity
primary site of expression of enzymatic defect is the bone marrow
resulting in accumulation and hyperexcretion of biologically inactive
bone marrow derived type I porphyrins which get distributed throughout
the body especially in urine, feces, blood and teeth. By virtue of their
phototoxicity these porphyrins accout for multiple pathologies of the
integument. Subepidermal bulous lesions progress to crusted erosions
which heal with scarring and either hyperpigmentation or rarely
hypopigmentation. Repeated damage from secondary infections may lead to
epidermal atrophy, pseudoscleroderma, resorption of distal phalanges and
severe functional limitations . Facial mutilation especially of the nose
and auricular cartilages and ectropion, keratoconjunctivitis and even
loss of vision may occur. Hypertrichosis and alopecia are common and
erythrodontia is virtually pathognomonic of CEP. Red colored urine may
be passed from early childhood. Patients may display signs and symptoms
of anemia usually hemolytic with splenomegaly and porphyrin rich
gallstones. The spleen is the major site for removal of damaged or
hemolysed erythrocytes and splenomegaly frequently observed in CEP is
secondary to this process. All CEP patients have increased plasma
turnover, erythrocytes exhibit polychromasia, poikilocyatosis,
anisocyatosis and basophilic stippling. Incteased reticulocytes and
normoblasts may be observed in peripheral circulation. Compensatory
expansion of hypertrophic bone marrow may lead to pathological
fractures, veretebral compression and collapse. Shortness of stature and
rarely osteolytic or sclerotic lesions of skeleton. Onset is in infancy
though adult CEP onset too has been very rarely observed. [1-6] The two
main differential diagnoses are hepatic erythropoietic porphyria and
erythropoietic porphyria in mild cases.
Treatment includes symptomatic measures in form of sun protection,
betacarotene, and also splenectomy for intractable. Activated charcoal
given by mouth is sometimes effective and Bone Marrow Transplantation is
a known cure. [1,2,15,17]
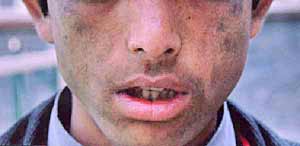
Fig. 1.
Our patient had a history of passage of red colored urine and
photosensitivity. Striking features of pinched nose, hypertrichosis of
face, with milia, scarring, hypo- and hyperpigmentation of photoexposed
parts, resorption of distal phalanges were observed. His teeth, urine,
stool and erythrocytes fluoresced intensely under Wood’s lamp. Increase
in plasma porphyrins a specific marker for cutaneous porphyrias was
observed. Out of cutaneous porphyrias Erythropoietic Protoporphyria was
ruled out as urinary porphyrins too are increased. Normal ALA and PBG
levels rule out acute porphyrias.The picture of increased urine, stool,
plasma and erythrocyte porphyrins on screening was highly suggestive of
CEP or a variant Hepato Erythropoietic Porphyria (HEP). [3,4,8,9,15,19]
 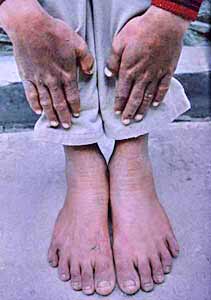
Fig 2 and Fig 3.
Our patient had greatly increased urinary free porphyrins fluorescing
intensely under UV light without extraction which again is a
characteristic feature of erythropoietic porphyrias [3-5,15].
Erythrodontia observed is pathognomonic of CEP (Fig 1-3). He had
hemolytic anemia with splenomegaly. For establishing a firm diagnosis,
quantitative porphyrin studies were used. Urinary Uroporpyrin levels
more than coproporphyrins, fecal coproporphyrin levels more than
uroporphyrins favor CEP. Erythrocytes having greatly increased levels of
coproporphyrins with small amounts of uroporphyrins and negligible
protoporphyrins again favoured CEP, as in HEP increased erythrocyte Zn-protoporphyrins
are an important feature. Also in HEP, the Zn-porphyrins coming in urine
need extraction for detection. [3,4,19]
Thus, with the above mentioned constellation of clinical, hematological
and biochemical features, a final diagnosis of puberty CEP onset was
entertained and is being reported in view of extreme rarity of this
condition.
|
|
| |
|
|
| |
REFERENCES
- Bickers DR,pathak MA,LIMHW. The Porphyrias .In :Fitzpatrick
TB,Eisen AZ,Wolff K .et al .(eds) pp1854-93.Dermatology in general
medicine . Mc Graw Hill Inc.New York :1993.
- Gupta S,Gupta U,Tiwari NK,Saraswat PK,Gupta DK. Gunthers disease
. Indian J Dermatol. 1998:43(2):79-81
- Kappas A, Sassa S,Galbriath RA , Nordmann Y pp. The porphyrias.
InThe metabolic and molecular basis of interited disease (eds):
Scriver C, Beaudett AL,Sly WS , Valle D .pp 2103-2139. 1995.
- Kauppinen R. Porphyrias .Lancet 2005 ; 365 (9463 ) :937-938.
- Poh- Fitzpatrick MB .The erythropoietic porphyrias. Dermotol
Clinical.1986; 4: 219-223.
- Nordman Y, Deyback JC .Congenital erythropoietic porphyria.
Semin. Liver Dis.1982; 2:154-158.
- Massod Q,Hassan I,Khan D,Sameen F,Quadri MI,Hussain ST, Majid S.
Congenital erythrooietic porphyria with hemolytic anemia . Indian J
Dermatol 2005;50 (3)155-157.
- Chatterji AK, Chatterjea JB. Porphyria Erythropoietica in India
. A review of 21 cases . J. Indian Med. Assos. 1977;56: 255-263.
- Kontos AP ,Ozog D, Bichakjian C, Lim HW. Congenital
erythropoietic porphyria associated with myelodysplasia presenting a
72 year old man : report of a case and review of literature.Br. J
Dermatol. 2003;148 (1) 160-4.
- THE MERCK MANUAL, Sec. 2, Ch. 14, The Porphyrias
- Rimington C (1971). Broadsheet 70 Association of Clinical
pathologists, London.
- Moore M R ( 1983). Broadsheet 109 Association of Clinical
Pathologists. London.
- Poh Fitzpatrick M.B : Laboratory testing in porphyrias. Int. Jl.
of Dermatol. 1979;18:453.
- Cripps DJ , Peters HA . Fluorescing erythrocytes and porphyria
screening test on urine, stool and blood . Arch. Dermatol . 1967;96
: 712-15.
- Poh Fitzpatrick MB. Erythropoietic Porphyria : Current
mechanistic , diagnostic and therapeutic considerations . Semin.
Hematol.1977 ;14 : 211-215.
- Majid S,Massod QA,Hassan I, Veshnavi S and Hussain ST .
Feasibility of diagnosis and typing of Porphyrias in developing
countries like India-its biochemical basis . JK Practitioner
,2006:13(1) 30-33.
- Tezcan, I., Xu, W., Gurgey, A., Tuncer, M., Cetin, M., Öner, C.,
Yetgin, S., Ersoy, F., Aizencang, G., Astrin, K.H., Desnick, R.J.
Congenital Erythropoietic Porphyria successfully Treated by
Allogeneic Bone Marrow Transplantation. Blood . 1998;92: 4053-4058.
- Mukherjee SK, Pimstone NR, Gandhi SN ,Tan KT . Biochemical
diagnosis and monitoring , therapeutic modulations of disease
activity in an unusual case of congenital erythropoietic porphyria .
Clin Chem . 1985;31 : 1946- 48.
- De Leova , Poh- Fitzpatrick MB, Mathew Roth MH , Marbel C .
Erythropoietic protoporphyria :ten year experience . Am. J. Med
.1976; : 60 : 8 -11.
|
|
| |
Corresponding Address:
Sabhiya Majid
Govt. Medical College Srinagar,
Jammu and Kashmir, India
Res : 0194 -2434214, Mob : 9419011275
email: sabumajid@yahoo.com
Paper received: 25.05.2006.
Paper accepted: 10.12.2006.
Published online: 31.01.2007. |
|
|
|
|
|
|
|
|
|
|
|
[ Home ] [ Gore/Up ][ <<< ] [ >>> ]
|
|
|
Infotrend Crea(c)tive Design |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|