| |
UVOD: OPŠTE OSOBINE INSULINA
Insulin (INS) je hormon endokrinog pankreasa i jedan je od
najznačajnijih proteina kičmenjaka. Povećana koncentracija glukoze (Glu)
u krvi stimuliše β ćelije pankreasa i dovodi do sekrecije INS
egzocitozom.
U krvi INS se nalazi u slobodnom obliku, odnosno ne postoje specifični
proteini plazme za koje se vezuje. Usled toga, njegov poluživot je
kratak (oko 5 min.) [1].
INS se vezuje za svoj stereospecifični receptor- receptor za insulin (IR)
koji se nalazi na plazma membrani velikog broja ćelija. Najveća
koncentracija IR nalazi se na ćelijama jetre, mišića, masnom tkivu i
limfocitima [1].
Pre svega, INS povećava transport Glu kroz membrane ćelija u većini
tkiva, a naročito u poprečnoprugastim mišićima, masnom tkivu i glatkim
mišićima [2]. INS favorizuje transport Glu difuzijom. Kada koncentracija
Glu u ćeliji premaši koncentraciju Glu u vanćelijskoj tečnosti, unos Glu
prestaje. U ćelijama se vrši fosforilacija Glu tako da ne ostaje gotovo
nimalo slobodne Glu, što omogućava unos novih količina u ćeliju. Osim na
transport Glu, deluje i na transport drugih monosaharida (ksiloza,
arabinoza, galaktoza) [1].
U nedostatku INS, transport Glu u većini ćelija smanjen je za jednu
četvrtinu normalne vrednosti, te tada tkiva u energetske svrhe koriste,
umesto Glu, masti i proteine. Izuzetak su mozak i srce(1).
INS utiče i na povećanje količine glikogena u skeletnim mišićima, koži,
žlezdama i dr. Na sličan način deluje i na jetru, ali se početni efekat
razlikuje od efekta u drugim tkivima. Na početku delovanja INS povećava
otpuštanje Glu iz hepatocita u krv, čime se količina glikogena u jetri
smanjuje. Deset časova od početka infuzije Glu, INS usmerava transport
Glu u suprotnom pravcu, tj. Glu tada ulazi u ćelije jetre i kao
posledica toga dolazi do povećanja količine glikogena u jetri.
Indirektno INS stimuliše lipogenezu, omogućavajući nastanak prekursora
za sintezu masnih kiselina, ali i direktno delujući na aktivaciju enzima
koji učestvuju u sintezi masnih kiselina i holesterola (1).
Na metabolizam belančevina INS deluje indirektno preko metabolizma
ugljenih hidrata, a u manjoj meri time što povećava transport
belančevina kroz ćelijske membrane. Povećavajući metabolizam ugljenih
hidrata, INS, utiče na racionalno korišćenje proteina (2). On smanjuje
katabolizam proteina u tkivima i zato dolazi do njihovog intenzivnijeg
anabolizma, čime INS posredno favorizuje rast i neophodan je za dejstvo
hormona rasta (1).
Takođe, INS, povećava transport fosfata i kalijuma u ćelije. Smatra se
da favorizuje transport fosfata tako što se u ćelijama stvara veća
količina glikozofosfata, čime se smanjuje koncentracija fosfatnih jona u
ćelijama, a to povoljno deluje na transport fosfata u ćelije. Glavna
mesta degradacije INS jesu jetra i bubrezi.
RECEPTOR ZA INSULIN
Receptor za insulin (IR) pripada grupi membranskih receptora koji
poseduju tirozin kinaznu aktivnost. Po strukturi je heterotetramerni
glikoprotein koji čine dve ekstraćelijske α i dve transmembranske β
subjedinice koje su najvećim delom locirane unutar ćelije α subjedinice,
kao i α i β subjedinice međusobno, povezane su disulfidnim vezama [2].
α subjedinica IR predstavlja njegovu regulatornu, ligand vezujuću
komponentu i poseduje region bogat cisteinom, koji je bitan za vezivanje
INS. Pri vezivanju liganda dolazi do konformacione promene koja
omogućava proces autofosforilacije β subjedinice [2, 3].
β subjedinica pretstavlja katalitičku subjedinicu IR i po funkciji spada
u tirozin (Tyr) specifične protein kinaze. Ekstraćelijskim delom
ostvaruje interakciju sa α subjedinicom, transmembranski deo učvršćuje
receptor u ćelijskoj membrani i učestvuje u njegovoj internalizaciji
nakon vezivanja hormona, a intraćelijski region uz membranu poseduje
regulatornu funkciju, jer sadrži Tyr ostatak na položaju 960 koji se
fosforiliše i bitan je za prepoznavanje ćelijskih supstrata. U
regulatornom regionu β subjedinice nalazi se i lizin na položaju 1018
koji učestvuje u vezivanju ATP-a, kao i tri Tyr ostatka na položajima
1146, 1150, 1151, koja pretstavljaju mesta autofosforilacije IR.
Fosforilacija ovih aminokiselina dovodi do konformacione promene koja
omogućava pristup ATP-u i ćelijskim supstratima koje receptor
fosforiliše. C-terminalni region β subjedinice receptora sadrži dva Tyr
ostatka (1316 i 1322) koje IR autofosforiliše i serin/treonin(Ser/Thr)
ostatke (1293, 1294, 1334), čija afosforilisanost reguliše kinaznu
aktivnost receptora. Nakon autofosforilacije (bar šest mesta), β
subjedinica je sposobna da fosoriliše i druge ćelijske supstrate [4].
INSULINSKI SIGNALNI PUT U SKELETNIM MIŠIĆIMA
Skeletni mišicći (SM) preuzimaju oko 85% ukupne Glu pri insulinskoj
stimulaciji [5,6] i Glu se u njih inkorporira i skladišti u vidu
glikogena [7].
Preuzimanje Glu u SM uglavnom zavisi od INS, mada može biti stimulisano
i nezavisno od INS i njegovog receptora, primenom fizičkog napora ili
električnih stimulacija[3].
Glu u ćelije ne ulazi procesom proste difuzije već uz pomoć proteina
nosača, koji olakšavaju transport Glu kroz ćelijsku membranu. Do sada je
iz sisarskih tkiva klonirano pet tipova proteina koji olakšavaju
transport Glu i identifikovani su kao glukozni transporteri (GLUT) od
GLUT1 do GLUT5 (3). Iako su sličnih struktura, svaki član GLUT familije
ima specifičnu distribuciju po tkivima i specifične regulatorne
mehanizme da bi se zadovoljile različite potrebe različitih tkiva [3].
U ćelijama SM, imunocitohemijskim metodama, lokalizovani su GLUT1 koji
je prisutan u većini tkiva, uključujući mozak i eritrocite i GLUT4 koji
je prisutan i u adipoznom tkivu [3, 8].
Ćelijska kultura humanih SM ćelija omogućuje odgovarajući model sistem u
kome se mogu proučavati molekularni mehanizmi insulinskog signalnog puta
[9]. Tako je primenom ove metode otkriveno da u mioblastima i miotubama
u kulturi dolazi do znatno manje ekspresije GLUT4, a da je znatno veća
ekspresija GLUT1 nego u intaktnim SM i interesantno je da se ekspresija
GLUT1 smanjivala sa diferencijacijom mišićnih ćelija u kulturi, a
paralelno sa redukcijom ekspresije GLUT1, redukovalo se i preuzimanje
Glu, i to kako pri bazalnim uslovima tako i pri stimulaciji INS [8,9].
U bazalnim uslovima GLUT su u ćeliji u vezikulama. U odgovoru na
stimulaciju INS dolazi do egzocitoze vezikula sa GLUT i GLUT1 i GLUT4
translociraju se na površinu membrane mišićne ćelije. Poremećaji u
mehanizmima koji su odgovorni za ovu translokaciju, vode insulinskoj
rezistenciji. Međutim, količina translociranog GLUT4 pri stimulaciji INS
je mnogo veća nego količina translociranog GLUT1. U SM ćelijama 90% Glu
transporta je posredovano GLUT4-om [10]. Čak je pokazano da u humanim i
mišićnim ćelijama pacova, transport Glu pri stimulaciji insulinom može u
potpunosti biti zadovoljen translokacijom samo GLUT4 na ćelijsku
membranu [3]. GLUT4 je u mogućnosti da uveća transport Glu pri
stimulaciji INS od 10 do 40 puta [10]. Nasuprot tome, uz pomoć GLUT1
ostvaruje se manji deo transporta Glu u mišićnim ćelijama, smatra se INS
nesenzitivnim, manjeg je afiniteta za Glu i vrši transport Glu u
bazalnim uslovima [10]. Dakle, INS stimulisano preuzimanje Glu u
mišićnim ćelijama posredovano je translokacijom GLUT4 na površinu
ćelijske membrane, dok GLUT1 posreduje u preuzimanju Glu u mišićne
ćelije pri bazalnim uslovima [8].
Signalni put iniciran INS, a koji vodi do translokacije GLUT4 na
ćelijsku membranu, uključuje interakcije autofosforilisanog IR (preko
Tyr960) sa fosfotirozin vezujućim domenima (PTB), kao i za tzv. PH
domenima ("pleckstrin homology") supstrata. PTB domeni predstavljaju
kratke oligopeptidne sekvence u strukturi proteina koje prepoznaju i
vezuju se za regione u proteinu u kojima se nalazi fosforilisani Tyr. PH
domeni takođe su kratke oligopeptidne sekvence u proteinima koje su
značajne za ćelijsku lokalizaciju proteina, a ime im potiče od
plekstrina, molekula u kome su otkrivene [4]. Dosadašnja istraživanja su
pokazala da već na nivou IR dolazi do divergencije INS signala. Identifikovano
je više ćelijskih proteina koje IR fosforiliše pri stimulaciji INS, te
je ta grupa proteina dobila ime supstrati insulinskog receptora (IRS)
[7].
U mišićnim ćelijama IR fosforiliše IRS-1, inače prvi otkriveni IRS i do
sada detaljno okarakterisan. Poseduje preko 20 karakterističnih sekvenci
sa Tyr koji može biti fosforilisan i više od 30 Ser/Thr sekvenci čija
fosforilacija igra ulogu u regulaciji funkcije IRS-1. IRS-1
protein-protein interakcijom prenosi signal na sledeće učesnike signalne
mreže. IRS-1 funkcioniše kao ”docking” protein, interagujući uglavnom
preko svojih regiona koji sadrže fosforilisane Tyr sa SH2 proteinima
[7]. SH2 proteini su dobili ime po SH2 domenima ("src-homology 2 domain")
koje sadrže i koji su otkriveni kod src proteina i koji interagju sa
regionima proteina u kojima se nalaze fosforilisani Tyr. Regulatorna
subjedinica fosfatidil inozitol 3 kinaze (PI-3K) je SH2 protein [4].
Tyr fosforilisani IRS-1 vezuje se za p85α regulatornu subjedinicu PI-3K
i p110 katalitička subjedinica biva aktivirana [11]. Ona fosforiliše
inozitolne molekule na položaju 3 i funkcioniše kao Ser kinaza.
Fosfatidil inozitolni (PI) molekuli se vezuju za protein kinazu B (PKB)
i aktiviraju je fosforilacijom na bar dva mesta (Thr i Ser) [7].
Aktivacija PKB je, po svemu sudeći, esencijalna za efikasan transport
Glu pri insulinskoj stimulaciji. Jednom aktivirana PKB fosforiliše GLUT4
i kinazu 3 glikogen sintaze (GSK-3) i inaktivira je. Inaktivacija GSK-3
potpomaže aktivaciju glikogen sintaze i povećava sintezu glikogena (polimer
glukoze, polisaharid). [12] Sinteza glikogena pretstavlja primarni put
anaerobnog metabolizma glukoze. [12]
Regulacija aktivnosti glikogen sintaze je veoma kompleksna i uključuje
fosforilaciju devet amino kiselina različitim kinazama i defosforilaciju
proteinskom fosfatazom-1 (PP-1). Povećana koncentracija glikogena
smanjuje aktivnost glikogen sintaze kako u prisustvu tako i u odsustvu
INS, jer je koncentracija glikogena u SM ograničena i postoji mehanizam
povratne sprege, a takodje, koncentracija glikogena utiče i na na
preuzimanje Glu u SM. [7]
INSULINSKA REZISTENCIJA I DIABETES MELLITUS TIP 2
Periferna insulinska resistencija koja uključuje poremećaje
transporta Glu u adipocitima i SM, ključni je faktor u patogenezi
Diabetesa Mellitusa tipa 2 (DMT2). [6]
DMT2 ili INS nezavisan oblik šećerne bolesti, je bolest koja uglavnom
nastaje sadejstvom genetičkih faktora i faktora spoljašnje sredine, mada
u nekim slučajevima, faktori spoljašnje sredine kao što su neaktivnost,
gojaznost i stres više doprinose nastanku oboljenja [5]. DMT2 je
prisutan u oko 90% svih dijabetičara i karakteriše ga periferna insulinska
rezistencija praćena deficitom u sekreciji INS. Da bi se razvio DMT2 oba
poremećaja moraju biti prisutna. [12]
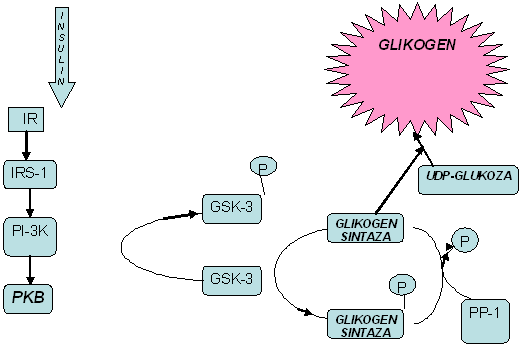
Šema br. 1: Aktivacija glikogen sintaze posredovana insulinom:
Vezivanje insulina za njegov receptor (IR), dovodi do aktivacije
fosfokinaze B (PKB). Supstrat PKB je kinaza 3 glikogen sintaze (GSK-3K)
koja biva inhibirana, čime je omogućena aktivnost glikogen sintaze i
sinteza glikogena.
Normalno, INS se vezuje za IR na efektornim ćelijama, što dovodi do
niza reakcija u ćeliji uz pomoć kojih se vrši transport Glu u ćeliju i
njeno metabolisanje. Insulinska rezistencija je nesposobnost da
periferna ciljna tkiva odgovore na odgovarajući način na fiziološku
koncentraciju INS prisutnu u cirkulaciji, stoga po definiciji insulinska
rezistencija je defekt u signalnoj transdukciji. [2] Da bi se postigle
fiziološke vrednosti glikemije, pankreas pojačano luči INS te dolazi do
kompenzacije insulinske rezistencije. Međutim, nakon perioda kompenzovane
insulinske rezistencije ipak dolazi do oštećenja tolerancije Glu iako se
koncentracija INS povećava sa povećanjem insulinske rezistencije.
Konačno, dolazi do poremećaja funkcije β ćelija i one ne uspevaju da
luče dovoljno INS. [12]
S obzirom na to da SM preuzimaju najveći deo ukupne Glu, insulinska
rezistencija u SM od velikog je značaja za razvoj DMT2 i može se javiti
na tri nivoa: na nivou transporta Glu, heksokinaze tj. fosforilacije Glu
i na nivou sinteze glikogena. [5,12]
S obzirom na to da je transport Glu jedan od prvih ograničavajućih
koraka u metabolizmu Glu, a nishodno je regulisan u DMT2-u, gen za GLUT4
bio je kandidat za prosvetljavanje molekularnih mehanizama uključenih u
perifernu insulinsku rezistenciju, međutim nije pronađena korelacija
između insulinske rezistencije i snižene ekspresije GLUT4 u DMT2, pošto
je snižena ekspresija GLUT4 prisutna i u nekih zdravih osoba, ali primećen
je poremećaj u transportu GLUT4 na površinu ćeliske membrane mišićne
ćelije pri insulinskoj stimulaciji. [5]
U pacijenata sa DMT2 poremećen transport Glu pri insulinskoj stimulaciji
u SM udružen je sa smanjenom količinom proteina IR i IRS-1, smanjenom
fosforilacijom Tyr u IR i IRS-1 i redukovanom aktivnošću PI-3K.
Redukovana Tyr kinazna aktivnost IR i redukovana fosforilacija IRS-1
nije povezana sa promenama u količini proteina IRS-1, pošto je
ekspresija IRS-1 ista u pacijenata sa DMT2 i zdravih osoba. [5]
Identifikovano je nekoliko polimorfizama u genu za IRS-1 i pronađeno je
da su prisutni kako u pacijenata sa DMT2, tako i u zdravih osoba, te se
za sada to ne smatra bitnim za nastanak DMT2. [5]
S obzirom na to da se smatra da je aktivacija PI-3K ključni korak u
preuzimanju Glu pri stimulaciji INS, rađeni su eksperimenti u kojima se
pratila aktivacija PI-3K pri stimulaciji INS u zdravih osoba i osoba sa
DMT2 i utvrđeno je da do povećane aktivnosti PI-3K pod uticajem INS u
DMT2 pacijenata ne dolazi. [5] Osim toga, u ovih pacijenata redukovana
je i aktivnost PKB [11]. Ipak, snižena aktivnost PI-3K pri stimulaciji
INS ne mora nužno da dovede i do redukcije u aktivciji PKB. Nađeno je
da INS dovodi do normalne fosforilace PKB u SM koji su rezistentni na
INS, uprkos u isto vreme smanjenju aktivnosti PI-3K [11].
U deksametazonom indukovanoj insulinskoj rezistenciji, iako ne dolazi do
poremećaja u fosforilaciji IR pri insulinskoj stimulaciji, dolazi do
inhibicije asocijacije IRS-1 i PI-3K i snižene fosforilacije PKB i GSK-3
i kompletno se blokira uticaj INS na defosforilaciju i aktivaciju
glikogen sintaze, a da pri tome nema uticaja na ekspresiju ovih proteina,
pri čemu farmakološki inhibirana GSK-3 u deksametazonom izazvanoj
insulinskoj rezistenciji dovodi do povećanja u sintezi glikogena, ali
pri tome nema poboljšanja u INS stimulisanom transportu Glu [11].
Transport Glu je osnovni korak u preuzimanju Glu u SM pri insulinskoj
stimulaciji i ograničavajući je faktor za celokupan metabolizam Glu u
organizmu. Da bi se utvrdila razlika između poremećaja u transportu Glu
i poremećaja u delovanju heksokinaze II u sintezi glikogena u mišićima
pri insulinskoj rezistenciji, meren je nivo slobodne Glu u ćeliji, jer
po transportu Glu u ćeliju ona biva fosforilisana uz pomoć heksokinaze u
glukozo-6-fosfat. Defekt u heksokinazi II anticipiran je pri
hiperinsulinemiji ako je više povišen nivo slobodne Glu u ćeliji u
odnosu na povećanje u nivou glukozo-6’fosfata, dok se poremećaj u
transportu Glu pripisivao ako je odnos povećanja nivoa bio
proporcionalan. [13]. Nekoliko grupa autora kloniralo je i
okarakterisalo gen za heksokinazu II i došli su do zaključka da mutacija
u genu za heksokinazu II ne doprinosi razvoju insulinske rezistencije i
DMT2 [5].
U pacijenata sa DMT2 INS stimulisano povećanje slobodne Glu je bilo
smanjeno ukazujući na to da je primarni defekt u transportu Glu pri
redukovanoj sintezi glikogena [13].
ZAKLJUČAK
SM preuzimaju oko 85% ukupne Glu pri insulinskoj stimulaciji, te
insulinska rezistencija u SM umnogome doprinosi postprandijalnoj
hiperglikemiji koja je glavni patogeni faktor mnogih kasnijih
komplikacija u pacijenata sa DMT2 i može biti na nivou transporta Glu,
njene fosforilacije i sinteze glikogena.
Transport Glu je osnovni korak u preuzimanju Glu u SM pri insulinskoj
stimulaciji i ograničavajući je faktor za celokupan metabolizam Glu u
organizmu.
U insulin rezistentnim stanjima, mnogi poremećaji u insulinskoj
signalnoj mreži su okarakterisani, iako još uvek nije jasno koji od tih
poremećaja je primarni.
S obzirom na to, mehanizmi koji leže u osnovi insulinskog signalnog puta
u SM, predstavljaju važno polje istraživanja pri proučavanju patofiziologije
DMT2 i insulinske rezistencije.
|
|
| |
LITERATURA
- Petrović,V. Cvijić,G. Endokrinologija. 1. izdanje, Beograd:
Zavod za udžbenike i nastavna sredstva; 1997.
- Pessin JE, Saltiel AR. Signaling pathways in insulin action:
molecular targets of insulin resistance. The Jurnal of clinical
Investigations 2000; Volume 106, Number 2: 165-169.
- Ryder JW, Chibalin AV, Zierath JR. Intracellular mechanisms
underlying increases in glucose uptake in response to insulin or
exercise in skeletal muscle. Acta Phisiol Scand 2001; 171, 249-257.
- Korićanac G. Osobine i sinteza insulinskih receptora pod
delovanjem glukokortikoida kod pacova različite starosti. Doktorska
disertacija, Biološki fakultet, Beograd, 2000.
- Zierath JR, Krook A, Wallberg-Henriksson H. Insulin action in
skeletal muscle from patients with NIDDM. Mol.and Cellular
Biochemistry 1998; 182: 153-160.
- Chowdhury HH, Jovsek M, Kreft M, Mars T, Zorec R , Grubic Z.
Insulin induced exocytosis in single, in vitro innervated human
muscle fibres: a new approach. Pflugers Arch- Eur J Physiol 2005;
450: 131-135.
- Jensen J, Jebens E, Brennesvik OE, Ruzzin J, Soos MA,
Engebretsen EML et al. Muscle glycogen inharmoniously regulates
glycogen synthase activity, glucose uptake and proximal insulin
signaling. AJP-Endocrinology and Metabilism 2006; 290: 154-162.
- Al-Khalili L, Cartee GD, Krook A. RNA interference-mediated
reduction in GLUT1 inhibits serum- induced glucose transport in
primary human skeletal muscle cells. Biochem and Biophysical
Research Communications 2003; 307: 127-132.
- Al-Khalili L, Chibalin AV, Kannisto K, Zhang BB, Permert J,
Holman GD et al. Insulin action in cultured human skeletal muscle
cells during differentiation: assessment of cell surface GLUT4 and
GLUT1 content. Cell. Mol. Life Sci. 2003; 60: 991-998.
- Brennan CL, Hoening M, Ferguson DC. GLUT4 but not GLUT1
expression decreases early in the development of feline obesity.
Domestic Animal Endocrinology 2004; 26: 291-301.
- Ruzin J, Wagman AS, Jensen J. Glucocorticoid-induced insulin
resistance in skeletal muscles: defects in insulin signaling and
effects of a selective glycogen synthase kinase-3 inhibitor.
Diabetologia 2005; 48: 2119-2130.
- Peterson KF, Schuman GI. Pathogenesis of skeletal muscle insulin
resistance in type 2 diabetes mellitus. The American Journal of
Cardiology 2002; 90: 11-18.
- Courtney HC, Olefskiy JM. Endocrinology. Insulin resistance.
Landes Bioscience, 2003.
www.eurekah.com
|
|
