|
Ateroskleroza je razbacani, čvorast oblik arterioskleroze, pojma kojim se obuhvataju sva vaskularna oboljenja sa zadebljanjem i otvrdnućem arterijskog zida. Primarno nastaje u intimi abdominalne aorte, njenim velikim i širokim ograncima, arterijama za donje ekstremitete, kao i koronarnim i cerebralnim arterijama (1). Karakteriše se akumulacijom ekstracelularnih i intracelularnih lipida, monocitno/makrofagnom infiltracijom, formiranjem penastih ćelija, proliferacijom glatkih mišićnih ćelija i akumulacijom vezivno-tkivnih proteina (2). Ranije opisivana kao degenerativna bolest, ateroskleroza se danas u literaturi opisuje sve češće kao hronična inflamatorna bolest, u čijem razvoju učestvuju humoralne i celularne imune reakcije.
Faktori koji podstiču aterogenezu
Po opšte prihvaćenoj teoriji o reakciji na oštećenje, proces ateroskleroze započinje oštećenjem endotelnih ćelija različitog stepena, koje može biti izazvano brojnim noksama. Mada je denudacija endotelnih ćelija inicijalno predložena kao prvi korak u aterogenezi, najnovija verzija ove hipoteze u prvi plan ističe endotelnu disfunkciju. Kao mogući uzroci endotelne disfunkcije navode se povišeni nivoi LDL čestica, kao i njihova modifikacija; slobodni radikali izazvani pušenjem cigareta, hipertenzijom i diabetes mellitus-om; genetske alteracije; povišene koncentracije homocisteina u plazmi; infektivni mikroorganizmi kao što su Chlamydia pneumoniae i cytomegalovirus; ili kombinacije ovih i drugih faktora (1,3)
Endotelna disfunkcija koja rezultira iz oštećenja praćena je kompenzatornim odgovorima koji menjaju normalne homeostatske osobine endotela. Adhezivnost endotela u odnosu na leukocite i trombocite se povećava, kao i endotelna permeabilnost. Oštećenje takođe indukuje ispoljavanje prokoagulantnih, umesto antokoagulantnih osobina endotela i obrazovanje vazoaktivnih molekula, citokina i faktora rasta. Ukoliko inflamatorni odgovor nije efikasan u neutralizaciji i uklanjanju uzročnih agenasa, on se nastavlja bez ograničenja, stimulišući migraciju glatkih mišićnih ćelija (GMĆ) u područje inflamacije i njihovu proliferaciju, te dovodeći do zadebljanja arterijskog zida. Međutim, zahvaljujući fenomenu "remodeliranja", zadebljanje zida se kompenzuje postepenom dilatacijom krvnog suda, tako da je u ovoj fazi lumen arterije još uvek nepromenjen. Granulociti su retko prisutni tokom bilo koje faze aterogeneze.Umesto toga, inflamatorni odgovor je posredovan makrofagima koji potiču iz monocita i specifičnim podtipovima T ćelija (3).
Kontinuirana inflamacija rezultira daljim povećanjem broja makrofaga i limfocita, koji migriraju iz krvi, umnožavaju se unutar lezija i oslobađaju hidrolitičke enzime, citokine, hemokine i faktore rasta koji doprinose daljem oštećenju. Nagomilavanje holesterola u makrofazima i glatkim mišićnim ćelijama vremenom uzrokuje nastanak morfoloških promena, tzv masnih tačaka i pruga na zidovima krvnih sudova. Ove promene tek neznatno sužavaju lumen, ne uzrokuju kliničke simptome i reverzibilne su ukoliko se obnovi integritet endotela (1). Međutim, ponavljana ili hronična oštećenja endotela će dovesti do daljeg nagomilavanja glatkih mišićnih ćelija, makrofaga, lipida i vezivnog tkiva u intimi. Glatke mišićne ćelije sintetišu mukopolisaharide, kolagen i elastin, a makrofazi propadaju i iz njih izlaze faktori koji podstiču proliferaciju glatkih mišićnih ćelija i fibroblasta, toksični lipidi oksidirani u makrofazima i litički lizozomalni enzimi, koji doprinose napredovanju ateroskleroze. Aterosklerotične lezije se smatraju uznapredovalim kada je akumulacija lipida, ćelija i komponenata matriksa udružena sa strukturalnom dezorganizacijom, reparacijom i zadebljanjem intime, kao i deformitetom arterijskog zida (1,4).
Hiperholesterolemija i modifikovani lipidi i lipoproteini
Hiperholesterolemija udružena sa povišenim nivoima aterogenih lipoproteina LDL i VLDL u krvi, vodi ka hroničnom prisustvu LDL čestica u arterijskom zidu. Ove LDL čestice mogu biti modifikovane oksidacijom, glikozilacijom (u dijabetesu), agregacijom, udruživanjem sa proteoglikanima ili inkorporacijom u imune komplekse i smatraju se glavnim uzrokom oštećenja endotela i glatkih mišića koji leže ispod endotela (3). Njihova uloga u širenju inflamatornog odgovora, u krajnjoj liniji se pripisuje stimulaciji replikacije makrofaga i podsticanju ulaska novih monocita na mesto lezije.
Kada LDL partikule penetriraju u intimu arterije, one bivaju zarobljene od glikozaminoglikana i izložene slobodnim radikalima iz susednih ćelija(5). Oksidativna modifikacija LDL-a je ekstremno kompleksna, jer obuhvata oksidaciju svih klasa lipida (sterola, masnih kiselina u fosfolipidima, holesterolskih estara i triglicerida) i proteinskih čestica(6). Povezana je sa formiranjem brojnih visokoreaktivnih molekula, kao što su lipidni peroksidi, lyso-PC, oksisteroli, i aldehidi, koji interakcijom sa susednim ćelijama, mogu uzrokovati nespecifičnu aktivaciju pri nižim koncentracijama ili ćelijsku smrt pri višim koncentracijama (5). Time se objašnjava uloga oksidirane LDL (oxLDL) u sledu događaja, koji obuhvata aktivaciju endotelnih ćelija, privlačenje monocita iz cirkulacije, adherenciju za endotelne ćelije i prodor u subendotelne slojeve zida, gde se diferenciraju u makrofage i eventualno, postaju penaste ćelije. Sama oxLDL je hemotaktički agens za monocite i T ćelije, koje se redovno nalaze u aterosklerotičnim lezijama(6). Povećana adhezija monocita za vaskularni endotel uslovljena je dejstvom oxLDL na oslobađanje hemotaktičkih faktora kao što su MCP-1 (monocyte chemoattracant protein-1) i M-CSF (macrophage colony-stimulating factor) , trombocitnog aktivirajućeg faktora -PAF (platelet -activating factor), leukotriena, kao i ekspresijom leukocitnih adhezivnih molekula i receptora za PAF (6,7). Ostali proaterogeni efekti oxLDL na endotelne ćelije uključuju indukciju ekspresije faktora rasta za glatke mišiće, indukciju formiranja superoksid anjona i apoptozu endotelnih ćelija. Humani endotelni receptor koji posreduje preuzimanje oxLDL pripada C tipu familije lektina -LOX-1 (lectinlike oxLDL receptor-1) i nedavno je kloniran (2).
Sposobnost oxLDL da indukuje akumulaciju holesterola u makrofagima bila je njegova prva opisana proaterogena osobina i poslužila je kao osnova za hipotezu da oksidacija LDL-a može biti važan korak u procesu aterogeneze. Naime, oksidativno modifikovane LDL čestice u arterijskom zidu, vezuju se za specifična mesta na površini makrofaga, koja su identifikovana kao "scavenger" receptori klase A (SRA) i koja posreduju u preuzimanju i degradaciji izmenjenih LDL čestica (8). Uklanjanje i sekvestracija modifikovanih LDL čestica su važni delovi inicijalne, protektivne uloge makrofaga u inflamatornom odgovoru, čime se minimiziraju efekti LDL na endotelne i glatke mišićne ćelije. Međutim, makrofagi ne poseduju regulatorne mehanizme nadzora nad ulaskom holesterola iz LDL čestica, pa postaju prepunjeni holesterolskim estrima i pretvaraju se u tzv penaste ćelije (1).
Utvrđeno je da oxLDL čestica poseduje mitogena svojstva i zahvaljujući tome, sposobna je da indukuje rast makrofaga. Nakon preuzimanja oxLDL posredstvom SRA, nastaje aktivacija protein-kinaze C i sledstvena sekrecija GM-CSF (granulocyte-monocyte colony-stimulating factor).U aterosklerotičnim lezijama, ova GM-CSF produkcija može igrati važnu ulogu u pripremi rasta makrofaga, u saradnji sa ostalim citokinima.Pored ove proliferativne uloge, nedavno je pokazano da SRA posreduju u rezistenciji makofaga na apoptozu. Isto tako, demonstrirano je da faktori koji stimulišu kolonije ( M-CSF i GM-CSF) i oxLDL podstiču ekspresiju PPAR-? (peroxisome proliferator-activated receptor- ?) u primarnim makrofagima i monocitnim ćelijskim linijama. Aktivacija PPAR-? (koji inače modifikuje aktivnost makrofaga) inhibira aktivaciju NF-?B i stoga, redukuje inflamatorni odgovor (8).
Sa druge strane, nedvosmisleno je utvrđeno da inflamatorni odgovor duboko utiče na kretanje lipoproteina unutar arterijskog zida. Medijatori inflamacije, kao što su TNF? (tumor necrosis factor ?) , IL-1 (interleukin-1) i M-CSF povećavaju vezivanje LDL za endotel i glatke mišićne ćelije i povećavaju transkripciju gena za LDL-receptore. Posle vezivanja za scavenger receptore in vitro, modifikovana LDL inicira seriju intracelularnih događaja, koji uključuju indukciju urokinaze i inflamatornih citokina, kao što je IL-1. Na taj način, zahvaljujući prisustvu ovih lipida, u arteriji se može održavati začarani krug inflamacije, modifikacije lipoproteina i dalje inflamacije (3).
Homocistein
Odranije je poznato da bolesnici sa homozigotnim defektom enzima neophodnih za metabolizam homocisteina, kao što su cistation beta sintetaza ili metilentetrahidrofolat reduktaza, još u detinjstvu razvijaju teške aterosklerotične promene, a mnogi od njih doživljavaju prvi infarkt miokarda već u dobi od 20 godina. Homocistein je toksičan za endotel, deluje protrombotički, povećava produkciju kolagena i smanjuje raspoloživost azot oksida, što objašnjava povezanost visokih koncentracija homocisteina u plazmi sa uznapredovalom aterosklerozom. Međutim, blago povišene koncentracije homocisteina u plazmi su pronađene i kod mnogih ljudi bez enzimskih defekata u metabolizmu homociteina. Utvrđeno je da ovi bolesnici imaju povećani rizik od simptomatske ateroskleroze koronarnih, perifernih i cerebralnih arterija (3).
Hipertenzija
Koncentracija angiotenzina II, koji je glavni produkt sistema renin-angiotenzin, često je povećana kod bolesnika sa hipertenzijom; angiotenzin II je snažan vazokonstriktor.Pored toga što uzrokuje hipertenziju, doprinosi aterogenezi stimulacijom rasta glatkih mišića. Angiotenzin II se veže za specifične receptore na glatkim mišićima, što rezultira aktivacijom fosfolipaze C i praćeno je povećanjem intracelularne koncentracije kalcijuma, kontrakcijom glatkih mišića, povećanom sintezom proteina i hipertrofijom glatkih mišića. Takođe se povećava lipoksigenazna aktivnost u glatkim mišićima, koja može povećati inflamaciju i oksidaciju LDL. Hipertenzija takođe ima proinflamatorna dejstva, jer povećava formiranje vodonik-peroksida i slobodnih radikala kao što su superoksid anjon i hidroksilni radikali u plazmi. Ove supstance redukuju produkciju azot-oksida u endotelu, povećavaju leukocitnu adheziju i povećavaju perifernu rezistenciju. Na taj način, formiranje slobodnih radikala posreduje izvesne efekte i hipertenzije i hiperholesterolemije (3).
Infekcija
Nedavni napredak u bazičnoj nauci, ustanovio je fundamentalnu ulogu inflamacije u posredovanju svih faza ateroskleroze, od inicijacije procesa, preko progresije do superponiranih trombotičkih komplikacija. Zato se oštećenje krvnog suda i pridruženi inflamatorni odgovor na oštećenje, danas navode kao esencijalne komponente aterogeneze. Međutim, "okidači" koji iniciraju i održavaju inflamatorni proces, još uvek nisu sa sigurnošću identifikovani. Mnogi istraživači veruju da oksidisani LDL i heat -shock proteins (HSPs) mogu izazvati inflamatorni odgovor i doprineti tinjajućoj inflamaciji u aterosklerotičnom plaku Pojedini autori razmatraju mogućnost autoimunog oštećenja zida krvnog suda antitelima koja se razvijaju kao odgovor na prisustvo ovih proteina. Drugi potencijalni kandidat, koji je predmet rastućeg interesa zbog sposobnosti da indukuje i inflamatorni i autoimuni odgovor, jeste infekcija (9,10).
Činjenice koje govore u prilog kauzalne uloge infekcije u patogenezi ateroskleroze, zasnivaju se na dokazima o prisustvu infektivnih agenasa unutar zida aterosklerotičnog krvnog suda ( prvo, cytomegalovirus, kasnije Chlamydia pneumoniae) i na seroepidemiološkim studijama koje demonstriraju povezanost patogen-specifičnih IgG antitela i ateroskleroze (CMV, Herpes simplex virus tip 1 i 2, C. pneumoniae, Helicobacter pylori, hepatitis A virus)(11). Moguće je da prisustvo infektivnog agensa (CMV i Chlamydia pneumoniae) u aterosklerotičnoj leziji, nastaje usled direktne infekcije zida krvnog suda patogenom koji nakon toga perzistira u latentnom stanju, produkujući abortivnu infekciju ili se replicirajući na niskom nivou. Alternativna mogućnost je da mijelomonociti čine rezervoar virusa, pri čemu cirkulišući monociti deluju kao vektori koji dopremaju viruse do mesta vaskularnog oštećenja ili inflamacije.
Dodatni dokazi u prilog kauzalnosti, demonstriraju uticaj infektivnih agenasa na celularne i molekularne promene koje pogoduju razvoju ateroskleroze. Tako, pojedini infektivni agensi (CMV) mogu stimulisati celularnu proliferaciju inhibicijom tumorskog supresorskog gena p53 ili povećanom sekrecijom faktora rasta, odnosno povećanom ekspresijom receptora za faktore rasta. Takođe, CMV infekcija inhibira apoptozu u humanim endotelnim ćelijama, povećava migraciju GMĆ iz medije i adventicije u neointimu i doprinosi akumulaciji lipida, povećanim preuzimanjem oksidisane LDL preko klase A scavenger receptora(10). Infektivni agensi mogu sudelovati u razvoju endotelne disfunkcije, promenom fenotipa endotelnih ćelija iz normalnog antikoagulantnog u prokoagulantni i pogoršanjem endotel-zavisnog vazodilatatornog odgovora (12). Konačno, CMV infekcija u GMĆ vodi razvoju slobodnih kiseoničnih radikala i može pojačati ekspresiju citokina, hemokina i adhezivnih molekula na površini ćelije (10).
Infekcija ne mora učestvovati u patogenezi ateroskleroze mehanizmima koji zahtevaju lokalno prisustvo patogena u zidu krvnog suda. Naime, infekcija izaziva i čitav spektar sistemskih efekata, uključujući promene u cirkulatornom nivou citokina, reaktanata akutne faze, belih krvnih zrnaca i odgovora posredovanih imunim sistemom, kao što je povećanje antitela usmereno na invaziju patogena. Različiti su mehanizmi putem kojih sistemski inflamatorni odgovor može ispoljiti efekte na oštećeni zid krvnog suda.
Molekularna mimikrija se predlaže kao mehanizam odgovoran za razvoj autoimunih bolesti i rezultira u pokretanju imunog odgovora usmerenog na sopstvene proteine. Koncept molekularne mimikrije zahteva infekciju patogenima koji sadrže homologe sekvence peptida sa proteinima domaćina. U tom slučaju, imuni odgovor, mada pokrenut invazijom patogena i usmeren na antigene patogena, može unakrsno reagovati i sa tkivima domaćina, koja pokazuju homologiju u aminokiselinskim sekvencama sa stranim antigenom. Npr, neke studije su povezale povećanu prevalencu periodontalne bolesti sa povećanom prevalencom ateroskleroze. Kod bolesnika sa gingivitisom , pronađen je značajno povišen nivo specifičnih pljuvačnih IgA antitela protiv HSP65 iz mikobakterija. Međutim, HSP65 iz mikobakterija je visoko homolog sa humanim HSP60. Istraživači su pretpostavili da stimulisana produkcija IgA antitela na unakrsno reaktivni antigen može doprineti gingivitisu, kao što može doprineti autoimuno-indukovanom aterosklerotičnom procesu (11).
Različiti cirkulišući faktori koji potiču iz patogena ili su stimulisani patogenima, mogu izazvati promene u monocitima/makrofagima koje mogu biti proaterogene ili mogu čak doprineti nestabilnosti plaka. Tako, lipopolisaharidi koji potiču iz bakterija i HSP(humani ili iz chlamydia) indukuju sekreciju tkivnog necrosis factora-? iz makrofaga , citokina koji stimuliše ekspresiju adhezivnih molekula na endotelnim ćelijama. Ovi cirkulišući faktori takođe stimulišu ekspresiju interleukina (IL)-1, mRNA i IL-6 u endotelnim ćelijama i glatkim mišićnim ćelijama i preferencijalno stimulišu endotelne ćelije da eksprimiraju E-selectin i ICAM-1 (intercelullar adhesion molecule-1). Svi pomenuti citokini: TNF-?, IL-1, IL-6 i ICAM-1 su povezani u aterogenezi. Pored toga, lipopolisaharidi i HSP takođe stimulišu sekreciju matrix metaloproteinaza, koje zahvaljujući kapacitetu da razgrađuju vezivno tkivo, mogu doprineti rupturi plaka (10).
Mada se povišeni cirkulišući nivo C-reaktivnog proteina (CRP) obično posmatra kao sistemski marker inflamacije, neke studije su ispitivale moguću kauzalnu ulogu ovog reaktanta akutne faze u aterogenezi. Npr, demonstrirano je da se CRP lokalizuje u blizini terminalnog kompleksa komplementa u intimi ranih humanih aterosklerotičnih lezija, što je dovelo do pretpostavke da CRP aktivira komplement i da aktivacija komplementa implicira u razvoju aterosklerotičnog plaka. Direktni proaterosklerotični i proinflamatorni efekat CRP-a je bar delimično posredovan endotelinom-1 i interleukinom-6 i ispoljava se povećanom ekspresijom ICAM-1, VCAM-1 (vascular-cell adhesion molecule-1) i E-selektina, povećanom produkcijom MCP-1 i povećanim preuzimanjem LDL u makrofagima (13,14). Direktna protrombotička uloga CRP-a u patogenezi koronarne ateroskleroze i njenim akutnim komplikacijama, ogleda se u indukciji monocitno/makrofagnog tkivnog faktora (TF)(15).
Iako su mnoge epidemiološke studije do sada pokazale da je seropozitivnost na izvesne infektivne agense povezana sa povećanim rizikom od kardiovaskularne aterosklerotične bolesti, može se zapaziti da rezultati različitih studija nisu bili uvek konzistentni. Moguće objašnjenje ove diskrepance bi bilo da doprinos prethodne infekcije kardiovaskularnoj aterosklerotičnoj bolesti zavisi od kapaciteta domaćina da suprimira inflamatornu aktivnost patogena. U prilog ove pretpostavke govore sledeće činjenice: a) neke osobe seropozitivne na CMV imaju povećane nivoe CRP-a, ali kod drugih je CRP unutar normalnog ranga. b) najviša prevalenca kardovaskularne aterosklerotične bolesti pojavljuje se u podgrupi sa kombinovanom CMV seropozitivnošću i povišenim nivoom CRP-a.Drugo moguće objašnjenje za diskrepancu u seroepidemiološkim studijama je povezano sa tipom imunog odgovora domaćina na infekciju, u kom slučaju bi humoralni ili celularni imuni odgovor određivao da li će određeni patogen doprineti razvoju aterosklerotične lezije kod određenog domaćina (10).
Inflamatorni procesi u ranoj fazi aterogeneze
Interakcija između endotelnih ćelija, monocita i T ćelija
Na pojedinim delovima arterija kao što su ogranci, bifurkacije i krivine, nastaju karakteristične promene u krvnom protoku koje se manifestuju smanjenim hemodinamskim stresom i povećanom turbulencijom. U ovim područjima, na endotelu se formiraju specifični molekuli odgovorni za adherenciju, migraciju i akumulaciju monocita i T-ćelija. Adhezivni molekuli na endotelnim ćelijama obuhvataju nekoliko selektina, intercelularne adhezivne molekule (ICAM) i vaskularne ćelijske adhezivne molekule (VCAM), koji funkcionišu kao receptori za integrine i glikokonjugate na monocitima i T ćelijama. Aktivacija monocita i T-ćelija vodi up-regulaciji receptora na njihovoj površini, kao što su mucin-like molekuli koji vežu selektine, integrini koji vežu adhezivne molekule iz superfamilije imunoglobulina i receptori koji vežu hemotaktične molekule. Ove ligand-receptor interakcije dalje aktiviraju mononuklearne ćelije, indukuju ćelijsku proliferaciju i pomažu u definisanju i lokalizaciji inflamatornog odgovora na mestu lezije. Adherencija monocita i T ćelija se može pojaviti posle povećanja u jednom ili više adhezivnih molekula, koji mogu delovati sinergično sa hemotaktičnim molekulima, kao što su MCP-1, osteopontin, IL-8 ili modifikovane LDL čestice (3).
Značajna je uloga koju sekretorne GMĆ imaju u mononuklearnoj ćelijskoj infiltraciji arterijskog zida. Kada se aktiviraju putem citokina, kao što je TNF-?, endotelne ćelije ispoljavaju specijalizovane adhezivne receptore [E- i P-selektine i vaskularni ćelijski adhezivni molekul-1 (VCAM-1)], čija eksperesija nije zabeležena u zdravim humanim arterijama. Sekretorne GMĆ generišu nekoliko agenasa koji mogu podstaći mononuklearnu ćelijsku infiltraciju u arterijski zid. One oslobađaju potentne hemotaktične faktore, kao što su IL-8 i MCP-1, i proinflamatorne citokine kao što su TNF-? i IL-1, koji indukuju ekspresiju adhezivnih molekula i leukocitnih hemotaktičnih faktora na endotelnim ćelijama. Nedavno je utvrđeno je da kokultura GMĆ i endotelnih ćelija, priprema endotelne ćelije, povećavajući njihovu osetljivost na proinflamatorne citokine, kao što je TNF-?, koji može biti prisutan u sistemskoj cirkulaciji u relativno niskoj koncentraciji. Priprema endotelnih ćelija se pripisuje citokinu TGF-ß (transforming growth factor ß), koji se proteolitički aktivira u biološki aktivnu formu putem serinske proteaze plazmina. Parakrina interakcija između GMĆ i endotelnih ćelija ima potencijal da amplifikuje regrutovanje leukocita ka mestu ustanovljenog ateroma i da egzacerbira inflamatorni proces koji leži u osnovi progresije bolesti (16).
Glatke mišićne ćelije - migracija i proliferacija
Rano u genezi aterosklerotičnog plaka, glatke mišićne ćelije (GMĆ) u mediji, podvrgnute mitogenoj stimulaciji, menjaju fenotip, gube kontraktilne elemente i stiču sposobnost da se repliciraju i migriraju unutar arterijskog zida. One vrše invaziju intime, gde doprinose progresivnoj hiperplaziji intime usled ekscesivne replikacije i taloženja fibroznog vezivno-tkivnog matriksa. Neointimalna hiperplazija, podstaknuta abnormalnom proliferacijom i migracijom GMĆ, posredovana je kompleksnom interakcijom brojnih faktora rasta, citokina, lipoproteina i lipida. Faktori koji se oslobađaju iz trombocita, ali i citokini oslobođeni iz hroničnih inflamatornih ćelija, uključujući T ćelije i makrofage, koji su prisutni u svim fazama aterogeneze mogu stimulisati oslobađanje citokina i faktora rasta iz istih ili obližnjih ćelija i na taj način igrati ključnu ulogu u aterogenezi (17).
Bazični fibroblastni faktor rasta (bFGF- basic fibroblast growth factor),npr, može inicirati proliferaciju GMĆ, dok trombocitni faktor rasta ( PDGF- platelet derived-growth factor) može uzrokovati posledičnu migraciju GMĆ ka intimi(18) Pokazano je da lisofosfatidilholin (lyso-PC), polarni fosfolipid koji se akumulira u oksidativno modifikovanoj LDL može indukovati migraciju GMĆ verovatno u kombinaciji sa PDGF ili ET-1 i da lyso-PC indukuje migraciju GMĆ bar delimično usled oslobađanja endogenog bFGF. Konverzija fosfatidilholina u lyso-PC tokom oksidativne modifikacije u arterijskom zidu može doprineti koronarnoj aterogenezi. (19).
Intimalna proliferacija i akumulacija matriksa pojavljuje se pod uticajem PDGF, TGF-ß, angiotenzina II i/ IGF-1 (insuline-like growth factor-1) (18). TGF-ß stimuliše preuzimanje L-arginina u glatkim mišićnim ćelijama i usmerava njegov metabolizam ka L-ornitinu, što rezultira produkcijom stimulatora rasta poliamina i integralnog konstituenta kolagena L-prolina (20). IGF osovina ispoljava dejstvo na aterosklerozu kroz efekte na rast GMĆ, migraciju i sintezu ekstracelularnog matriksa u aterosklerotičom plaku (21), dok je defekt u protektivnom mehanizmu IGF osovine odgovoran za povećanu apoptozu GMĆ i nestabilnost plaka (22). Angiotenzin II verovatno posredstvom mitogen-aktivirane proteinske kinaze izaziva rast i migraciju GMĆ (23). Proinflamatorni citokini IL-1ß, IL-4, IL-8 mogu takođe indukovati migraciju i proliferaciju GMĆ stimulacijom lipoksigenaznog puta metabolizma arahidonske kiseline (17). Gubitak inhibitora faktora rasta, kao što su azot-oksid iz endotelnih ćelija i heparan-sulfat proteoglikan, takođe može doprineti migraciji i proliferaciji glatkih mišićnih ćelija posle oštećenja (18).
Danas se s pravom ističe da je hronična inflamacija arterijskog zida osnovna karakteristika ateroskleroze, dok se citokini smatraju modulatorima inflamatornih događaja koji se pojavljuju tokom svih faza ateroskleroze. S obzirom da su GMĆ najzastupljenije ćelije aterosklerotičnog tkiva, pretpostavilo se da, pored leukocita, one mogu biti značajan izvor citokina u arterijskom zidu.Poznato je da neki faktori vezani za aterosklerozu pojačavaju produkciju citokina u glatkim mišićnim ćelijama, npr oksidativno modifikovani LDL ili trombin. Nedavno je demonstrirana sposobnost angiotenzina da izazove inflamatorni odgovor u GMĆ stimulacijom produkcije citokina i aktivacijom nuklearnog faktora ?B (24). Sličnu sposobnost pokazao je 5-hidroksitriptamin (serotonin) povećavajući sintezu IL-6, bar delimično putem protein kinaze C, u kulturi glatkih mišićnih ćelija. Interleukin-6 je multifunkcionalni proinflamatorni citokin, koji se proizvodi u GMĆ i izaziva njihovu kontrakciju i proliferaciju. Pored toga, IL-6 utiče na druge komponente aterosklerotične lezije, monocite/makrofage i endotelne ćelije, indukcijom izvesnih faktora, kao što su vaskularni endotelni faktor rasta (VEGF - vascular endothelial growth factor) i monocitni hemotaktični protein -1 (MCP-1) (25). IL-6 je moćan stimulator reakcije akutne faze, važan aktivator limfocita i induktor produkcije kolagena i glikozaminoglikana u fibroblastima. Produkcija IL-6 koristi se kao marker proinflamatornog potencijala glatkih mišićnih ćelija (24).
Monociti i imunitet
Adherencija monocita za promenjene endotelne ćelije i njihova transendotelna migracija u arterijski zid, gde ostaju u obliku makrofaga, verovatno su najranije ćelijske abnormalnosti u aterogenezi.
U procesu regrutovanja i migracije monocita na mesto inflamacije, hemotaksa nesumnjivo igra važnu ulogu i do sada je opisano više molekula koji ispoljavaju hemotaktičnu aktivnost na monocite: komponenta komplementa C5a, leukotrien B4, TNF-?, 12-hidroksieikosatetraenoična kiselina i monocitni hemotaktični protein-1 (MCP-1). MCP-1 se produkuje u različitim ćelijama arterijskog zida ( endotelnim ćelijama, fibroblastima i GMĆ). Stimulusi za produkciju MCP-1 uključuju minimalno modifikovane LDL čestice, i citokine kao što su IL-1, TNF, INF-? i M-CSF (26). Pored snažne hemotaktičke aktivnosti, podsticanja transmigracije i emigracije cirkulišućih monocita u tkivo, MCP-1 ispoljava i druge efekte na monocite, uključujući indukciju anjona superoksida i ekspresiju različitih proinflamatornih gena. Ekspresija MCP-1 na leukocitima, predominantno makrofagima, uticala je na povećanje broja makrofaga i dalje potenciranje imunog odgovora, akumulacijom oksidiranih lipida i ekspresijom integrina (27).
Makrofagi su stožerne ćelije u inflamatornom procesu udruženom sa tkivnim oštećenjem, pre svega zbog potenciranja inflamatornog odgovora usled produkcije različitih inflamatornih medijatora. Oni su "čistači" i antigen-prezentujuće ćelije; sekretuju hemokine, citokine (kao što su TNF-?, IL-1 i TGF-ß), faktore koji regulišu rast (kao što je PDGF, TGF- ß, bFGF i IGF-1), reaktivne kiseonične radikale i proteolitičke enzime (naročito metaloproteinaze). Sami monociti podstiču dodatnu adherencu monocita i migraciju iz vaskularnog prostora na mesto inflamacije (27).Visok afinitet IGF receptora, tip I, na površini makrofaga omogućava da sIGF modulira koncentraciju makrofaga na mestu oštećenja. Humani makrofagi takođe sintetišu i sekretuju IGF-I i neke od vezujućih proteina (IGF BP). IGF-1 sekretovan unutar aterosklerotične lezije je važan za monocitnu hemotaksu, aktivaciju i oslobađanje citokina (tj, TNF-?). Verovatno je da IGF koji potiče iz makrofaga povećava preuzimanje i degradaciju LDL, kao i brzinu esterifikacije holesterola u makrofagima (21).
U početku se mislilo da su jedine ćelije koje proliferišu tokom ekspanzije aterosklerotične lezije glatke mišićne ćelije. Međutim, replikacija makrofaga poreklom iz monocita i T ćelija je verovatno od jednake važnosti. Kontinuirani ulazak, preživljavanje i replikacija mononuklearnih ćelija u leziji zavisi delimično od faktora kao što su M-CSF i GM-CSF za monocite, a IL-2 za limfocite. Kontinuirano izlaganje M-CSF-u dozvoljava makrofagima preživljavanje in vitro i omogućava umnožavanje unutar lezije. Nasuprot tome, inflamatorni citokini kao što su INF-? pod izvesnim uslovima deluju na monocite, indukujući programiranu ćelijsku smrt (apoptozu). Ako se ovo dešava in vivo makrofagi bivaju uključeni u nekrotičku srž, karakterističnu za uznapredovale, komplikovane lezije (3).
Aktivirani makrofagi eksprimiraju antigene klase II histokompatibilnosti kao što je HLA-DR koji im omogućava da prezentuju antigene T limfocitima. S obzirom da su CD4 i CD8 ćelije prisutne u lezijama u svim fazama procesa, razumljiva je pretpostavka da su ćelijski posredovani imuni odgovori uključeni u aterogenezu. T ćelije se aktiviraju kada vežu antigen, koji je prethodno obrađen i prezentovan od strane makrofaga. Aktivacija T ćelija rezultira u sekreciji citokina, uključujući INF-? i TNF ? i ß, koji amplifikuju inflamatorni odgovor. Glatke mišićne ćelije iz lezije takođe poseduju klasu II HLA molekula na svojoj površini, verovatno indukovanu interferonom-gama i mogu takođe prezentovati antigene T ćelijama. Jedan od potencijalnih antigena je oksidisana LDL čestica, koju mogu produkovati makrofagi. Heat-shock protein 60 može takođe doprineti autoimunosti. Ovaj i drugi heat-shock proteini vrše nekoliko funkcija, uključujući nakupljanje, intracelularni transport, razgradnju proteina i prevenciju denaturacije proteina. Oni mogu biti povišeni na endotelnim ćelijama i učestvovati u imunom odgovoru (3).
Jedan imunoregulatorni molekul, CD40 ligand, može biti eksprimiran na makrofagima, T ćelijama, endotelu i glatkim mišićnim ćelijama u aterosklerotičnim lezijama in vivo. Njegov receptor CD40 je eksprimiran na istim ćelijama. Povećane koncentracije ovih molekula predstavljaju sledeći dokaz imune aktivacije u aterosklerotičnim lezijama. Dalje, ligand CD40 indukuje oslobađanje IL-1ß iz vaskularnih ćelija, potencijalno povećavajući inflamatorni odgovor. Inhibicija CD40 sa blokirajućim antitelima redukuje formiranje lezija u apolipoprotein E deficitarnim miševima (3).
Trombociti
Adhezija trombocita i muralna tromboza su uvek prisutne u iniciranju, kao i generisanju aterosklerotičnih lezija u ljudi i životinja. Trombociti mogu adherirati za disfunkcionalni endotel i mogu biti izloženi subendotelnom kolagenu i makrofagima. Kada se aktiviraju, trombociti oslobađaju svoje granule koje sadrže citokine i faktore rasta, a oni zajedno sa trombinom mogu doprineti migraciji i proliferaciji glatkih mišićnih ćelija i monocita. Aktivacija trombocita vodi formiranju slobodne arahidonske kiseline, koja može biti transformisana u prostaglandine, kao i tromboksan A2, jedan od najmoćnijih vazokonstriktornih i trombocit-agregirajućih supstanci koje su poznate, ili u leukotriene, koji mogu amplifikovati inflamatorni odgovor.
Ruptura plaka i tromboza su zabeležene komplikacije uznapredovalih lezija koje vode nestabilnom koronarnom sindromu ili infarktu miokarda. Trombociti su važni u održavanju vaskularnog integriteta u odsustvu oštećenja i zaštiti protiv spontane hemoragije. Aktivirani trombociti se mogu akumulirati na zidu arterije i regrutovati dodatne trombocite u širenju tromba. Važna komponenta trombocita je glikoprotein IIb/IIIa koji pripada integrin superfamiliji adhezivnih molekula-receptora i pojavljuje se na površini trombocita tokom trombocitne aktivacije i formiranja tromba. Ovi receptori imaju važne hemostatske funkcije i njihovi antagonisti preveniraju formiranje tromba kod bolesnika koji su imali infarkt miokarda (3).
Uznapredovale lezije ateroskleroze i prateće komplikacije
Prva i najkarakterističnija lezija uznapredovale ateroskleroze je aterosklerotični plak; on se sastoji od lipidima-bogate srži u centralnom delu ekscentrično zadebljale intime. Površinu plaka okrenutu lumenu pokriva fibrozna kapa (28). Centralni deo plaka je ispunjen žitkim kašastim sadržajem, koji nastaje povećanjem i konfluiranjem malih kolekcija ekstracelularnih lipida. Lipidi (pretežno LDL) dospevaju u krvni prostor direktno, insudacijom iz plazme, ili mogu biti endocitovani od strane makrofaga, putem scavenger receptora nakon oksidativne modifikacije i akumulirani indirektno posle nekroze makrofaga prepunjenih lipidima (29).
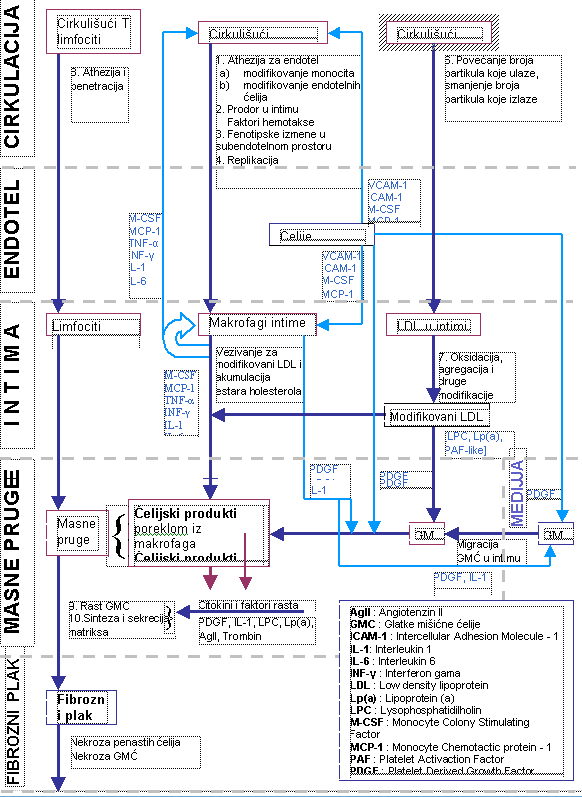
Shema 1. (prethodna strana): Ćelijske interakcije u toku aterogeneze; uloga različitih medijatora |
|
|
Vremenom se u plaku povećava i sadržaj vezivnog tkiva, koje se sastoji pretežno od kolagena i glatkih mišićnih ćelija. Sloj intime bogat proteoglikanima, zajedno sa makrofagima, glatkim mišićnim ćelijama, limfocitima i mast ćelijama biva potisnut u region između lipidne srži i endotelne površine. Lipidna srž je, naročito na lateralnim ivicama i sa luminalne strane, ograničena novoformiranim kapilarima (4).
Nestabilnost i ruptura plaka
U zrelim plakovima, zastupljene su u različitom odnosu dve glavne komponente plaka: mekana, lipidima bogata ateromatozna kaša i čvrsto, kolagenom bogato sklerotično tkivo. Sklerozna komponenta je obično daleko voluminoznija, stabilizuje plak i štiti ga od disrupcije, dok ateromatozna kaša destabilizuje plak i čini ga podložnim rupturi. Glavne determinante vulnerabilnosti plaka su veličina i sastav ateromatozne srži, debljina fibrozne kape koja pokriva plak, akutna inflamacija unutar kape i zamor kape, usled opterećenja. Predilekcono mesto za rupturu plaka je rubni region, gde je fibrozna kapa najtanja i infiltrisana makrofagima. Dezintegracija fibrozne kape je praćena iznenadnim izlaganjem visokotrombogene kaše protoku krvi(29).
Smatra se da rupturi plaka znatno doprinosi pogoršana sinteza kolagena. U humanom ateromu dokazano je prisustvo aktiviranih T ćelija, koje su prisutne u većem broju zajedno sa makrofagima na mestu rupture. Postoje dokazi da hronična imuna stimulacija unutar ateroma vodi produkciji INF-? iz T-ćelija, koji inhibira sintezu kolagena u vulnerabilnom regionima fibrozne kape. Produkcija INF-? se takođe povezuje sa inhibicijom proliferacije i povećanom sklonošću ka apoptozi u glatkim mišićnim ćelijama, kao i sa aktivacijom makrofagnih funkcija koje su odgovorne za vulnerabilnost plaka (28).
Makrofagi su sposobni da degradiraju ekstracelularni matriks fagocitozom ili sekrecijom proteolitičkih enzima kao što su plazminogen aktivatori i familija matriks metaloproteinaza (kolagenaze, želatinaze i stromelizini), koje mogu oslabiti fibroznu kapu, predisponirajući je za rupturu. Međutim, ekspresija kolagenaze, želatinaze B i stromelizina nesumnjivo je dokazana i u ostalim ćelijama - makrofagima, T-ćelijama i GMĆ humanog ateromatoznog plaka, kao i pojačana matriks-degradirajuća aktivnost u vulnerabilnim delovima plaka (28) Matriks metaloproteinaze (MMP) se luče u latentnoj zimogen formi i zahtevaju ekstracelularnu aktivaciju, posle čega su sposobne da degradiraju prividno sve komponente ekstracelularnog matriksa. U normalnim uslovima, aktivnost ovih enzima drže pod kontrolom sveprisutni tkivni inhibitori metaloproteinaza, dok aktivirane T ćelije mogu stimulisati produkciju metaloproteinaza u makrofagima, čime se podstiče nestabilnost plaka. Aktivirane mast ćelije, prisutne u malom broju u rubnom regionu, mogu da sekretuju moćne proteolitičke enzime, kao što su triptaze i himaze, koje učestvuju u aktivaciji pro-MMP. Neutrofili su takođe sposobni da razaraju tkivo sekrecijom proteolitičkih enzima, ali su retko prisutni u intaktnim plakovima (29).
Ostale komplikacije
Na prethodno rupturiranom plaku ili intaktnom plaku, može se javiti iznenadna tromboza usled promena u funkciji trombocita, koagulaciji i/ili fibrinolizi, što je verovatno važan mehanizam odgovoran za potpunu okluziju krvnog suda i pojavu infarkta. Štetna uloga makrofaga posle rupture plaka ogleda se u podsticanju generisanja trombina i luminalne tromboze putem tkivnog faktora (28,29). Tkivni faktor je integralni membranski protein, koji se veže za faktor VII/VIIa i inicira koagulacionu kaskadu. Eksprimiran je na površini lipidima-prepunjenih monocita, makrofaga i penastih ćelija u humanim aterosklerotičnim lezijama. Ekspresija tkivnog faktora može biti promptno indukovana lipopolisaharidima, kao i inflamatornim medijatorom koji se nalazi u aterosklerotičnim lezijama, CD40L, dok aktivatori PPAR?, člana nuklearne familije receptora, redukuju ekspresiju i aktivnost tkivnog faktora (30).
Ruptura plaka je takođe praćena razvojem hematoma kroz rascep fibrozne kape, mada neki hematomi mogu nastati unutar lezije zbog krvarenja iz novoformiranih krvnih sudova. Zreli plakovi, naročito ako sadrže veću količinu fibroznog vezivnog tkiva, mogu kalcifikovati, pri čemu mineralni depoziti zamenjuju izumrle ćelije i ekstracelularne lipide. Uznapredovale lezije su često udružene sa lokalizovanom dilatacijom dela krvnog suda koji zauzimaju.
Ovako nastale aneurizme mogu sadržati muralne trombe, a delići tromba koji se otkidaju u sistemsku cirkulaciju mogu dovesti do embolije. Čak i u odsustvu komplikovanih lezija, sa napredovanjem ateroskleroze sve više se sužava lumen krvnog suda, remeti se protok krvi i raste rizik od daljeg oštećenja endotelnih ćelija; time se zatvara začarani krug. Suženje lumena krvnog suda praćeno je tkivnom hipoksijom, a zbog smanjene elastičnosti krvnih sudova povećava se krvni pritisak. Time se povećava mogućnost rupture izmenjenog zida krvnog suda i posledičnog izlivanja krvi u okolno tkivo, naročito u uslovima naglog i većeg porasta krvnog pritiska. Različiti tipovi lezija pokazuju korelaciju sa određenim kliničkim sindromima (4,1).
Značaj novih saznanja
Ćelije mogu eksprimovati različite konstelacije gena i stoga varirati fenotipski , zavisno od njihovog okruženja. Nove tehnike razvijene za identifikaciju DNA bi trebale doneti ogromnu količinu informacija o genima koji se eksprimuju i načinu njihove ekspresije , što bi pomoglo rasvetljavanju kompleksne prirode aterogeneze (31). Kako je ateroskleroza poligenska bolest, razumevanje načina genske ekspresije bi verovatno moglo bar delimično objasniti razlike u prijemčivosti za agense koji uzrokuju bolest. Osim toga, način genske ekspresije može varirati u lezijama različitih osoba i na različitim mestima i može nam pružiti indicije u pogledu genetskih razlika koji se tiču ne samo prijemčivosti već i odgovora na terapiju.
Napredak u molekularnoj genetici je omogućio uklanjanje ili inserciju gena i determinisanje uloge njihovih proizvoda u bolesti (32). Proizvedeni su brojni animalni modelu koji se upotrebljavaju u studiranju genetike aterogeneze, kao što su apolipoprotein E-deficijentni miševi (33). U odsustvu apolipoproteina E, ostaci lipoproteina se ne nose u jetru gde se normalno metaboliziraju, pa miševi postaju hiperholesterolemični i razvijaju aterosklerotične lezije slične humanim. Studije na transgeničnim miševima su otkrile da Lp(a), holesterol estar transfer protein, apolipoprotein A (glavni apoprotein high-density lipoproteina), i slični molekuli imaju mali efekat na aterogenezu, dok je M-CSF (macrophage colony-stimulating factor) izgleda važan u regulaciji brojnih monocita i makrofaga i formiranju lezija (33,34).
Prema tome, mada je hiperholesterolemija važna u približno 50% bolesnika sa kardiovaskularnom bolešću (35), uvek treba razmatrati i ulogu drugih faktora. Danas je sve jasnije da je ateroskleroza inflamatorna bolest, a ne rezultat proste akumulacije lipida. Kada bismo mogli selektivno modifikovati štetne komponente inflamacije u arterijama, a zadržati intaktne zaštitne mehanizme, mogli bismo kreirati i nove prilaze dijagnostici i vođenju bolesti kod onih 50% bolesnika sa kardiovaskularnom bolešću koji nemaju
hiperholesterolemiju.
Literatura
- Bierman EL: Atherosclerosis and the other forms of arteriosclerosis in: Braunwald E, Isselbacher KJ, Wilson JD et al. Harrison?s Principles of Internal medicine. 13 th edition
McGraw-Hill.Inc 1995
- Morawietz H, Rueckschloss U, Nieman B, Duerrschmidt N, Galle J, Hakim K, Zerkowski HR, Sawamura T, Holtz J: Angiotensin II induces LOX-1, the Human Endothelial Receptor for Oxidized Low-Density Lipoprotein. Circulation 1999; 100: 899-902
- Ross R: Atherosclerosis - An Inflammatory Disease. The New England Journal of Medicine 1999; 340(2): 115-126
- Stary HC, Chandler B, Dinsmore RE, Fuster V, Glagov S, Insull W, Rosenfeld ME, Schwartz CJ, Wanger WD, Wissler RW: A Definition of Advanced Types of Atherosclerotic Lesions and a Histological Classification of Atherosclerosis. Circulation 1995; 92: 1355-1374
- Jovinge S, Ares MPS, Kallin B, Nilsson J: Human Monocytes/Macrophages Release TNF-? in response to Ox-LDL. Arteriosclerosis, Thrombosis and Vascular Biology 1996; 16: 1573-1579
- Steinberg D: Lewis A. Conner Memorial Lecture. Oxidative modification of LDL and Atherogenesis. Circulation 1997; 95 (4): 1062-1071
- Mehta A, Yang B, Khan S, Hendricks JB, Stephen C, Mehta JL: Oxidized Low-Density Lipoproteins Facilitate Leukocite Adhesion to Aortic Intima Without Affecting Endothelium-Dependent Relaxation. Arteriosclerosis, Thrombosis and Vascular Biology 1995; 15: 2076-2083
- de Winther MPJ, van Dijk KW, Havekes LM, Hofker MH: Macrophage Scavenger Receptor Class A. Arteriosclerosis, Thrombosis, and Vascular Biology 2000; 20:290-302
- Libby P, Ridker PM, Maseri A: Inflammation and atherosclerosis. Circulation 2002;105: 1135-1143
- Epstein SE, Zhou YF, Zhu J: Infection and atherosclerosis: Emerging Mechanistic Paradigms. Circulation 1999; 100: e20-e28
- Mayr M, Kiechl S, Willeit J, Wick G, Xu Q: Infections, Immunity, and Atherosclerosis: Associations of Antibodies to Chlamydia pneumoniae, Helicobacter pylori, and Cytomegalovirus With Immune Reactions to Heat-Shock Protein 60 and Carotid or Femoral Atherosclerosis. Circulation 2000; 102 (8): 833-841
- Liuba P, Karnani P, Pesonen E, Paakkari I, Forslid A, Johansson L, Persson K, Wadstrom T, Laurini R: Endothelial Dysfunction After Repeated Chlamydia pneumoniae Infection in Apolipoprotein E-Knockout Mice. Circulation 2000; 102(9): 1039-1048
- Verma S, Li SH, Badiwala MV, Weisel RD, Fedak PWM, Li RK, Dhillon B, Mickle DAG: Endothelin Antagonism and Interleukin-6 Inhibition Attenuate the Proatherogenic Effects of C-Reactive Protein. Circulation 2002; 105(16): 1890-1896
- Pasceri V, Willerson JT, Yeh ET: Direct Proinflammatory Effect of C-Reactive Protein on Human Endothelial Cells. Circulation 2000; 102(18): 2165-2171
- Nagacomi A, Freedman B, Geczy CL: Interferon-? and Lipopolysaccharide Potentiate Monocyte Tissue Factor Induction by C-Reactive Protein: Relationship With Age, Sex, and Hormone Replacement Treatment. Circulation 2000; 101 (15): 1785-1791
- Rainger GE, Nash GB: Cellular Pathology of Atherosclerosis: Smooth Muscle Cells Prime Cocultured Endothelial Cells for Enhanced Lekocite Adhesion. Circulation Researsh 2001; 88: 615-627
- Natarajan R, Rosdahl J, Gonzales N, Bai W: Regulation of 12-Lipoxygenase Cytokines in Vascular Smooth Muscle Cells. Hypertension 1997; 30: 873-879
- Braun-Dullaeus RC, Mann MJ, Dzau VJ: Cell Cycle Progression. New Therapeutic Target for Vascular Proliferative Disease. Circulation 1998; 98: 82-89
- Kohno M, Yokokawa K, Yasunari K, Minami M, Kano H, Hanehira T, Yoshikawa J: Induction by Lysophosphatidylcholine, a Major Phospholipid Component of Atherogenic Lipoproteins, of Human Coronary Artery Smooth Muscle Cell Migration. Circulation 1998; 353-359
- Durante W, Liao L, Reyna SV, Peyton KJ, Schafer AI: Transforming Growth
Factor-ß1 Stimulates L-arginine Transport and Metabolism in Vascular Smooth Muscle Cells: Role in Polyamine and Collagen Synthesis. Circulation 2001; 103: 1121
- Bayes-Genis A, Conover CA, Schwartz RS: The Insulin-Like Growth Factor Axis. A review of Atherosclerosis and Restenosis. Circulation Research 2000; 86: 125-135
- Patel VA, Zhang QJ, Siddle MA, Soos MA, Goddard M, Weissberg PL, Bennett MR: Defect in Insulin-Like Growth Factor-1 Survival Mechanism in Atherosclerotic Plaque-Derived Vascular Smooth Muscle Cells Is Mediated by Reduced Surface Binding and Signaling. Circulation Research 2001; 88: 895-907
- Xi XP, Graf K, Goetze S, Fleck E, Hsueh WA, Law RE: Central Role of the MAPK Pathway in Ang II-Mediated DNA Synthesis and Migration in Rat Vascular Smooth Muscle Cells. Arteriosclerosis, Thrombosis and Vascular Biology 1999; 19: 73-82.
- Kranzhofer R, Schmidt J, Pfeiffer CAH, Hagl S, Libby P, Kubler W: Angiotensin Induces Inflammatory Activation of Human Vascular Smooth Muscle Cells. Arteriosclerosis, Thrpombosis and Vascular Biology 1999; 19: 1623-1629
- Ito T, Ikeda U, Shimpo M, Yamamoto K, Shimada K: Serotonin Increases Interleukin-6 Synthesis in Human Vascular Smooth Muscle Cells. Circulation 2000; 102: 2522-2532
- Takahashi M, Masuyama JI, Ikeda U, Kasahara T, Kitagawa SI, Takahashi YI, Shimada K, Kano S: Induction of Monocyte Chemoattractant Protein-1 Synthesis in Human Monocytes During Transendothelial Migration In Vitro. Circulation Research 1995; 76: 750-757
- Aiello RJ, Bourassa PAK, Lindsey S, Weng W, Natoli E, Rollins BJ, Milos PM: Monocyte Chemoattractant Protein-1 Accelerates Atherosclerosis in Apolipoprotein E-Deficient Mice. Arteriosclerosis, Thrombosis and Vascular Biology 1999; 19: 1518-1525
- Libby P: Molecular Bases of the Acute Coronary Syndromes. Circulation 1995; 91: 2844-2850
- Falk E, Shah PK, Fuster V: Coronary Plaque Disruption. Circulation 1995; 92: 657-671
- Marx N, Mackman N, Schonbeck U, Yilmaz N, Hombach V, Libby P, Plutzky J: PPAR? Activators Inhibit Tissue Factor Expression and Activity in Human Monocytes. Circulation 2001; 103: 213-224
- Shalon D, Smith SJ, Brown PO. A DNA microarray system for analyzing complex DNA samples using two-color fluorescent probe hybridization. Genome Res 1996;6:639-645
- Chien KR. Genes and physiology: molecular physiology in genetically engineered animals. J Clin Invest 1996;97:901-909
- Nakashima Y, Plump AS, Raines EW, Breslow JL, Ross R. ApoE-deficient mice develop lesions of all phases of atherosclerosis throughout the arterial tree. Arterioscler Thromb 1994;14:133-140
- de Villiers WJS, Smith JD, Miyata M, Dansky HM, Darley E, Gordon S. Macrophage phenotype in mice deficient in both macrophage-colony-stimulating factor (op) and apolipoprotein E. Arterioscler Thromb Vasc Biol 1998;18:631-640.
- Braunwald E. Shattuck Lecture -- cardiovascular medicine at the turn of the millennium: triumphs, concerns, and opportunities. N Engl J Med 1997;337:1360-1369.
Adresa autora:
Biserka Tirmenštajn-Janković
Naselje Kraljevica C2 / II stan 13
19000 Zaječar
e-mail: tribally@ptt.yu
Rad je primljen: 23. 12. 2002.
Rad je prihvaćen: 20. 02. 2003. |
|