| |
UVOD
Alpotrov sindrom (AS) je hereditarna nefropatija, koja nastaje zbog
poremećaja u građi glomerulske bazalne membrane (GBM), koja je troslojne
strukture,sastavljena od dve lamine rare i lamine dense, debljine
310-380 nm. Kolagen IV čini strukturnu potku, a u sastav bazalne
membrane učestvuju još nidogen, proteoglikan i laminin. Postoji oko 14
podjedinica koagena tip IV. Molekul kolagena IV (COL4) je dug 400nm i
ima oko 6 lanaca, a nama su od važnosti lanci alfa-3 i 4 i alfa-5 lanac
(COL4A5) koji je mapiran na dugom kraku X hromozoma i čini lokus za X
vezani oblik Alportovog Sy (sastoji se od 51 eksona). U oko 70% obolelih
od Alportovog sindroma nedostaju COL4A3, COL4A4 i COL4A5 lanci. Najčešće
je nasleđivanje dominantno vezano za X hromozom (85%), ređe autosomno
recesivno i autosomno dominantno [1,2,5,7,9,10,17,20].
Prve podatke o udruženosti naslednog nefritisa i senzorineuralne gluvoće
dao je Alport.
Simptomi bolesti u klasičnim slučajevima javljaju se rano, obično oko 6.
godine života.
Za postavljanje dijagnoze ovog oboljenja treba da postoji trijas-pozitivna
porodična anamneza o postojanju hematurije i progresivan tok bolesti,
posebno kod osoba muškog pola, karakteristične patohistološke promene
pri elekromikroskopiji biopsijom uzetog i pregledanog bubrežnog tkiva,
audiometrijski nađena obostrana progresivna senzorineuralna nagluvost za
visoke tonove, tipične promene na očnom sočivu i retini (lentokonus,
katarakta) [1,2,3,5].
U zavisnosti od uzrasta kada se bolest ispoljila, postoji juvenilna i
adultna forma. Juvenilna forma se rano ispoljava,progresivnog je toka,
često odmah po rođenju se otkriva prisustvom hematurije, a hronična
bubrežna insuficijencija se razvija oko 20-te godine i kod svih obolelih
u porodici bolest ima sličan tok kao i vreme kada se razvija hronična bubrežna
insuficijencija (oko 30.-te godine života). Adultna forma je sporije
evolucije, a hronična insuficijencija bubrega se može razviti u 5-toj
deceniji života. Mikroskopska hematurija je prisutna skoro uvek kod
obolelih pacijenata muškog pola, već u neonatalnom uzrastu, a
recidivantna makrohematurija je prisutna u dobu do 15-te godine života u
trajanju i oko 10 dana a i duže udruženo sa proteinurijom koja može biti
i do 1gr/24h i loš je prognostički znak koji vodi u hroničnu bubrežnu
insuficijenciju. U oko 40% obolelih se razvija nefrotski sindrom. Kod
pacijenata ženskog pola mikrohematurija sa ili bez makrohematurije je
intermitentna i javlja se u oko 80-90% obolelih. Progrediranje u
hroničnu bubrešnu insuficijenciju je u 10-30% osoba ženskog pola i
javlja se u poznijim godinama života.
Oštećenje sluha je ekstrarenalana manifestacija Alportovog sindroma i
uvek je obostrano, progredijentno, senzorineuralnog tipa,najpre na visokim
tonovima, a kasnije zahvata sve frekvence. Ukoliko je juvenilna forma
bolesti, oštećenje sluha je izraženije, dok kod adultne forme je skoro
podjednako zastupljeno kod osoba oba pola i diskretniji je gubitak
sluha. Oštećenje sluha može biti prisutno i pre nego što postane
klinički vidljivo, pa je važan rani audiogram. Patohistološki, bazalna
membrana kohee, trpi isti vid promena kao i glomerulska bazilarna
membrana, zahvaćen je bazalni zavoj puža, usled defektne abnormalnosti u
sintezi kolagena IV, jednog od glavnih sastojaka bazanih membrana, što
je u slučaju X vezanog nasleđa mutacija gena na Xq22-24 (COL4A5).
Postoji klasična forma Alportovog sindroma koja nije praćena promenama u
sluhu. U nekom porodicama bubrežna bolest se javlja kasnije u odraslom
dobu i ne mora biti praćena gluvoćom [4,6,15,16,18,19].
Promene na očima, na sočivu (bazalna membrana sočiva) i retini
izraženije su kod juvenilne forme. Pojava lentikonusa je prisutna skoro
u ¼ obolelih, kada su i promene na retini skoro uvek prisutne, ali
obično bez simtoma. Mogu biti promene i u vidu keratokonusa,
mikrosferofakija..
U nekim slučajevima se mogu naći i trombocitopenija sa džinovskim
trombocitima abnormalne ultrastrukture i funkcije, mentalna retardacija
uz hiperproteinemiju, neurološki poreećaji (polineuropatija, Morbus
Charcot-Marie-Tooth), ihtioza, distrofična bulozna epidermoliza,
eofagealna, traheobronhijalna, genitalna lejomiomatoza, fotomionukleus,
dijabetes mellitus.
Elektronskom mikrografijom se registruje zade-bljanje GBM sa
raslojavanjem lamine dense uz prisustvo mikrogranula, kao tipičan
histološki nalaz. Kod dece promene su segmentne a kod odraslih difuzne
prirode.Kod dečaka su izraženije segmentne promene i povečavaju se u
korelaciji su sa intenzitetom proteinurije i uzrastom deteta [14,17].
S obzirom da je Alportov sindrom nasledna bolest kolagena IV, zavisno od
toka bolesti postoji najmanje 10 različitih fenotipova ove bolesti, što
se može objasniti postojanjem genske heterogenosti. Postoje opisi oko
100 mutacija gena koji kontrolišu sintezu alfa 5 lanca, pa je zato i
tolika fenotipska raznovrsnost AS [17,20,21].
Alportov sindrom je bolest loše prognoze. Muški pol, oštećen sluh,juvenilna
forma, promene na očima su loši prognistički znaci. Mutacija na genu
COL4A5, posebno delecije su loš prognostički parametar. Specifična
terapija za ovu bolest ne postoji.
Obolelim osobama sa terminalnom bubrežnom insuficijencijom treba
primeniti hemodijalizu ili uraditi tranplantaciju. Jedina svrishodna
terapija bi bila genska, jer bi se sprečio osnovni poremećaj [11,12,13].
CILJ RADA
Cilj rada je da se ukaže na značaj primene raspoloživih
dijagnostičkih metoda u otkrivanju ove sindromske nasledne bolesti, gde
je prvi siptom bila obostrana nagluvost dečaka, diferencijalno dijagnostički
razlučiti potencijalne izolovane bolesti uva, povrede, kongenitalna
oštećenja sluha, sindriomske bolesti, genetske i negenetske etiologije a
u cilju postavljanja prave dijagnoze i terapijskih opcija.
METOD RADA
Za prikaz bolesnika, podaci su dobijeni anamnezom o progresivnoj
obostranoj nagluvosti, kliničkim ORL pregledom, impendancmetrijom ,
tonalnom audiometrijom, OtoAkustičkom Emisijom, laboratorijskim nalizama
krvi i urina, histološkim nalazom biopsijom uzetog bubrežnog tkiva,
oftalmološkim pregledom.
REZULTATI RADA
Pacijent uzrasta 8 godina, muškog pola,osrednje osteomuskularne
građe, javio se na ORL pregled zbog obostrane progresivne nagluvosti
koja se javila par meseci ranije i postepeno pogoršavala. Klinički-otoskopski
pregled je bio uredan, timpanometrijom je dobijen Ty A obostrano, koheo-stapedijalni
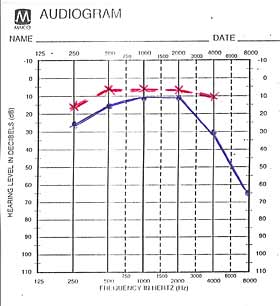
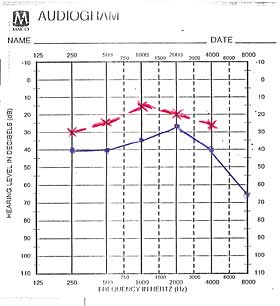
refleks je pokazao lako povišen prag sluha, audiološki zapis je pokazao
obostranu senzorineuralnu nagluvost na visokim frekvencama, izražajniju
na levom uvu, nishodnog tipa do 65dB, u početku za tzv ''govorne
frekvence'', potom i do 8000Hz.. Auditivni evocirani potencijali su potvrdili
da se ne radi o retrokohleranom oštećenju, a Otoakustička Emisija je
ostala bez odgovora kohlee, što je potvrdilo da je pad sluha veći od 40
dB. Svi parametri su bili u prilog senzorineuralnog oštećenja suha. U
ličnoj anamnezi, dete je rođeno prirodnim putem, u terminu, nije bilo
podataka o prenatalnim peri ili postnatalnim noksama, povredama, uredno
je vakcinisano, urednog psihomotornog razvoja. Redovno je pohađao školu
(sistematskim pregledom pre upisa u školu nije se registrovalo bilo koje
oštećenje koje bi pobudilo sumlju na ovu bolest). U porodičnoj anamnezi
nije bilo nagluvih, ali su deda po ocu i stric bili bubrežni bolesnici,
o čemu nije bilo dostupne dokumentacije. Roditelji su bili zdravi.
Laboratorijske analize, pre svega urin ukazali su na mikrohematuriju (++
i +++) i proteinuriju od 1gr-117mg /24h (analize ponavljane u roku od 10
dana), uz negativan builirubin i hemoglobin S- urea je bila 7,3 mml/L,S-kreatinin
je bio 121 µmol/L a hiperkalcijemija do 5mmol/L. KKS nije ukazivala na
anemiju a SE je bila 15mm/H. Pacijent je upućen na nefrološko
ispitivanje u referentnu ustanovu, gde je nakon biopsije tkiva bubrega (elekronskom
mikroskopijom nađena naizmenična istanjenja i zadebljanja glomerularne
bazalne membrane sa raslojavanjem, retikuliranjem i fragmentiranjem
lamine dense) postavljena dijagnoza Alportovog sindroma (AS). Zbog
pozitivne, iako nepotpune anamneze na hereditarnu sindromsku bolest,
roditelji deteta su podvrgnuti adekvatinim pregledima, koji su pokazali
da nisu oboleli od AS, te da se radilo o autozomnom recesivnom nasleđivanju.
Oftalmološkim pregledom je nađen lentikonus, na retini nije bilo
promena, a vid je bio uredan.Visina krvnog pritiska je bila u granicama,
adekvatna dobu pacijenta. Boja kože i vidljivih sluzokoža je bila bez
promena koloriteta.
DISKUSIJA
Hereditarni nefritis Alport je teška progresivna nasledna bolest,
incidence pojavljivanja 1-5000 do 1-10000, u oko 85-90% nasleđivanje je
autozomno dominantno, vezano za X hromozom, loše je prognoze, a
terapijska procedura je simptiomatska.
Ukoliko je u porodici definisana mutacija gena, moguća je prenatalna
dijagnoza. Imunohistohemijska ispitivanja kože i GBM sa antitelima na
COL4A5, COL4A3, COL4A4 su od velikog značaja, ali su izvodljiva u samo
za to opremljenim laboratorijama za genetski inženjering, što je u našim
uslovima još uvek u povoju. |
|
| |
ZAKLJUČAK
Alportov sindrom je teška nasledna sindomska bolest, praćena
nefritisom, senzorineuralnom gluvoćom i abnormalnostima oka,
progredijentnog je toka i loše prognoze.
U današnjim uslovima terapijska procedura je simptomatska, a genska
terapija, koja je u razvoju, bila bi jedino svrsishodna jer bi se
sprečila pojava bolesti, a ne samo kupiranje simptoma bolesti usled
prisutnih genski determinisanih oštećenja.
Za sada se sprovodi gensko savetovanje koje je značajno zbog
informisanja familije o prirodi bolesti i zbog pravovremenog
uključivanja dece oštećenog sluha u habilitacioni postupak. Genetsko
savetovanje je korisno jer daje prognozu o progrediranja nagluvosti
LITERATURA
- J Am Soc Nephrol. 2003 Jul;14(7):1794-803- Prakash S, Chung KW,
Sinha S, Barmada M, Chung KW, Sinha S, Barmada M, Ellis D, Ferrell
RE, Ferrell RE, Randhawa PS, Dinda A, Vats A - Autosomal dominant
progressive nephropathy with deafness: linkage to a new locus on
chromosome 11q24
- Klin Oczna. 2004;106(3):332-4 – Slowik M, Popiela G,
Kazimierczak K, Szelepin L, Popiela G, , Szalinski M. - Alport's
syndrome--case report
- Bol Med Hosp Infant Mex. 1993 Aug;50(8):596-602.- Garcia-Torres
R, Orozco L- Alport's syndrome: new finding
- Clin Otolaryngol Allied Sci. 1995 Apr;20(2):158-63- Sirimanna KS,
France E Stephens SD - Alport's syndrome: can carriers be identified
by audiometry?
- Medicine (Baltimore). 1999 Sep;78(5):338-60-Kashtan CE.- Alport
syndrome. An inherited disorder of renal, ocular, and cochlear
basement membranes
- Rev Bras Otorrinolaringol (Engl Ed). 2005 Nov- DecDec;71(6):813-9.
-Alves FR, de A Quintanilha Ribeiro F- Revision about hearing loss
in the Alport's syndrome, analyzing the clinical, genetic and bio-molecular
aspects
- Hum Mutat. 2006 Oct;27(10):1061.- King K, Flinter FA, Green PM.
- A two-tier approach to mutation detection in the COL4A5 gene for
Alport syndrome
- Pediatr Nephrol. 2007 May;22(5):621-5. Gubler MC.- Diagnosis of
Alport syndrome without biopsy?
- Am J Pathol. 1994 May;144(5):986-96.- Yoshioka K, Hino S,
Takemura T, Maki S, Wieslander J, Takekoshi Y, Makino H, Kagawa M,
Sado Y, Kashtan CE - Type IV collagen alpha 5 chain. Normal
distribution and abnormalities in X-linked Alport syndrome revealed
by monoclonal antibody
- Exp Biol Med (Maywood). 2007 May;232(5):638-42.- Wang Y, Zhang
H, Ding J, Wang F. Correlation between mRNA expression level of the
mutant COL4A5 gene and phenotypes of XLAS females.
- Kidney Int. 2004 May;65(5):1551-5 .-Imai E, Takabatake Y, Mizui
M, Isaka Y. Gene therapy in renal diseases
- Kidney International. 51(5):1493-9, 1997 May.- Tryggvason K.
Heikkila P. Pettersson E. Tibell A. Thorner P. Can Alport syndrome
be treated by gene therapy?
- Kidney International. 50(5):1445-63, 1996 Nov. / Kashtan CE.
Michael AF. Alport syndrom
- Am J Pathol. 2001 Sep;159(3):1097-104 .-Harvey SJ, Mount R, Sado
Y, Naito I,Ninomiya Y, Harrison R, Jefferson B, Jacobs R, Thorner PS.-
The inner ear of dogs with Xlinked nephritis provides clues to the
pathogenesis of hearing loss in X-linked Alport syndrom
- Nephrol Dial Transplant. 2001 Jun;16(6):1291-4.- Richardson D,
Shires M, Davison AMRenal diagnosis without renal biopsy. Nephritis
and sensorineural deafness.
- N Y State Dent J. 2007 Apr;73(3):34-7.- Friedman K, Velez I.
Alport syndrome. Report of a case with severe maxillofacial
manifestations.
- Laryngol Rhinol Otol (Stuttg). 1976 Jan;55(1):6-16. Weidauer H,
Arnold W. Morphological changes in the inner ear of alport's
syndrome (author's transl)
- Eur Arch Otorhinolaryngol. 1996;253(8):470-4Meyer zum
Gottesberge AM, Reuter A, Weiher H.- Inner ear defect similar to
Alport's syndrome in the glomerulosclerosis mouse model Mpv17.
- Curr Opin Neurol. 1996 Feb;9(1):3.- Baloh RW. Vestibular and
auditory disorders.
- Kidney Int. 1995 Apr;47(4):1142-7.-Gubler MC, Knebelmann B,
Beziau A, Broyer M, Pirson Y, Haddoum F, Autosomal recessive Alport
syndrome:immunohistochemical study of type IV collagen chain
distribution.
- J Am Acad Audiol. 1995 Jan;6(1):73-9. Wester DC, Atkin CL,
Gregory MC-Alport syndrome: clinical update.
- Alport syndrome: clinical update. -Keats BJ.. Genes and
syndromic hearing loss.
|
|