| |
|
|
INTRODUCTION
More than four decades ago, many countries initiated neonatal
screening programs in order to identify newborns with inherited
metabolic and endocrinological diseases for which early diagnosis
and treatment would prevent serious and permanent health disorders.
Phenylketonuria was the first disorder included in newborn screening
in many countries. In the decades after that, the program expanded
gradually, and included an increasing number of severe disorders
that result in a high degree of physical and intellectual
disability.
The World Health Organization defines the role of screening as the
detection of a treatable disease, with an adequately understood
natural history, in the asymptomatic phase, in order to initiate
treatment and prevent symptoms or to delay complications. Newborn
screening began to be applied in 1960 with the work of the American
microbiologist Dr. Robert Guthrie. The first international
discussion on newborn screening organized by the World Health
Organization was held in 1967 when a group of scientists on
congenital metabolic disorders discussed the technical and ethical
aspects of screening.
Guthrie's test is a mandatory health care measure and is performed
on every newborn, whether healthy or sick, born on or before the due
date. This laboratory analysis is usually performed already in the
maternity ward, most often in the first 48 hours after the baby's
birth, although it can be done up to the 8th day of the baby's life.
The current recommendation of the Advisory Committee on Inherited
Diseases in Infants and Children, the current version of which dates
from 2016 in the USA, defines a "recommended universal screening
panel" consisting of a basic list of 34 diseases and an expanded
list that includes 26 more diseases. Diseases for which screening is
recommended can be classified into several groups: organic acid
metabolism disorders, fatty acid oxidation disorders, amino acid
metabolism disorders, endocrine disorders and hemoglobinopathies.
From endocrine disorders, screening is recommended for congenital
hypothyroidism and congenital adrenal hypoplasia within the basic
panel [1]. The list of diseases that will be covered by the
screening test depends on the health system of the country and its
screening program. Which disease will be checked mostly depends on
its frequency, on the availability of therapy, but also on how
developed the country is and whether it has the means to pay for
screening for all newborns.
Neonatal screening for hypothyroidism has been introduced in
Montenegro since 2008 as a mandatory form of health care for
newborns, and it is the only disease from the group of hereditary
endocrinological diseases that screening includes.
Screening for phenylketonuria
Screening for phenylketonuria is a prerequisite for the early
application of a restricted diet, which is necessary for the
prevention of severe neurological disorders in children diagnosed
with the disease. Phenylketonuria is the most common congenital
metabolic disorder that causes a severe degree of physical and
mental disability if it is not diagnosed in a timely manner and
therapeutic treatment is not started. Phenylketonuria is a treatable
disease and is listed in the national newborn screening program in
countries around the world. Newborns with positive screening
indications can achieve a satisfactory therapeutic effect by timely
control of phenylalanine intake after diagnosis. The combination of
early diagnosis and initiation of treatment results in normal
physical and intellectual development for most children with
phenylketonuria. Phenylketonuria and other hyperphenylalaninemia are
a group of hereditary disorders that arise due to disorders in the
oxidation of the amino acid phenylalanine to tyrosine [2].
Phenylketonuria has a special place among hereditary metabolic
diseases. It is the first disease from that group in which the link
between a hereditary biochemical disorder and mental retardation was
clearly established (Fǿlling 1934), the first disease from that
category for which the possibility of dietary treatment was
discovered (Bickel 1954) and the first for which a laboratory test
was developed a test used in newborn screening in the entire newborn
population (Guthrie 1963) [3]. The prevalence of phenylketonuria in
the world is around 1: 10.000 newborns [4].
Phenylalanine is an essential amino acid, of which, after resorption
from the intestines, a smaller amount is incorporated into body
proteins, and the remaining, larger part must be oxidized into
tyrosine with the help of the enzyme phenylalanine-hydroxylase in
the liver. Phenylketonuria is caused by mutations in the gene
encoding the liver enzyme phenylalanine hydroxylase. The consequence
is enzyme insufficiency and the inability to oxidize phenylalanine
to tyrosine with an increase in the concentration of phenylalanine
and its "abnormal" metabolites in cells and body fluids. Today, the
mechanism by which phenylalanine or its metabolites in high
concentrations damage brain function is not yet known, but it is a
fact that maintaining them within normal limits in phenylketonuric
children with an appropriate dietary regimen prevents brain damage
[5].
Figure 1. A child with phenylketonuria
https://img.medscapestatic.com/pi/meds/ckb/07/44107tn.jpg
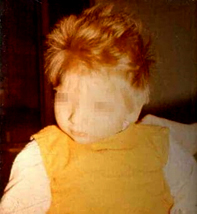
Children with classic phenylketonuria have no noticeable symptoms
in the first days and weeks of life. It is only after a few weeks
that signs of slowed psychomotor development appear, children do not
learn to walk, sit at the right time, 25% of children have epileptic
seizures, develop hypotonia of muscles, psychomotor restlessness,
behavioral changes, microcephaly, lag in physical development. About
a quarter of the affected children have infantile eczema,
hypopigmentation of the skin and hair, and a mouse-like smell of
sweat and urine. Severe mental retardation occurs already during the
first year (IQ 30) [6].
As every newborn is screened for phenylketonuria (Guthrie's test),
the concentration of phenylalanine and tyrosine in the blood is
determined in children with a positive Guthrie screening test. Based
on the value of phenylalanine in the blood, the disease is
classified as mild hyperphenylalaninemia: 120–360 mmol; light gray
zone 360–600 mmol; mild form of phenylketonuria: 600–900 mmol;
moderate: 900–1200 mmol and classical>1,200 mmol [7].
Treatment of phenylketonuria is carried out by lifelong restriction
of phenylalanine intake to the amount necessary for the construction
of own proteins from birth. In infants, milk formulas with little
phenylalanine are exclusively used. The implementation of the diet
has a threefold goal:
- The accumulation of an excessive amount of phenylalanine in
the blood (and therefore in the brain) is prevented by strict
control of the natural protein/phenylalanine intake.
- Replacing natural protein that has been removed from the
diet with a safe or phenylalanine-free protein, called a
synthetic protein, amino acid blend/supplement, or protein
replacement. All protein replacements are phenylalanine-free or
very low in phenylalanine.
- Achieving normal growth and nutritional status. This is
achieved by ensuring that the diet contains a balanced intake of
all nutrients and energy. Vitamin and mineral supplements are
either added to protein replacement or given as a separate
supplement.
In the diet, the intake of foods rich in phenylalanine is
restricted for life: milk, dairy products, meat, fish, chicken,
eggs, beans, nuts. The intake of fruits, vegetables and cereals is
recommended in the diet [8].
The prognosis of untreated phenylketonuria is poor considering the
deterioration of mental and nervous functions, the accompanying
symptomatic epilepsy and the difficulties and complications that
threaten such a child. About half of untreated children live to be
20 years old, and about a third live to be 30 years old. With timely
diagnosis at an early age and adequate dietary nutrition, children
with treated phenylketonuria do not differ from healthy peers.
Prevention begins before the birth of a child, when a pregnant woman
with phenylketonuria implements a diet without phenylalanine. If the
diet is not strict before conception and during pregnancy, damage to
the central nervous system of the fetus, congenital heart defects
and microcephaly will occur. After birth, the newborn is given a
Guthrie test.
A sample should be taken from every healthy, sick, term and non-term
newborn. The exact period for sampling should not be less than 48
hours of protein feeding and should not exceed 30 days from birth;
however, the ideal period would be between the third and seventh day
of birth in newborns [9].
Since antibiotic therapy can make the test for phenylketonuria
falsely negative, the sample is generally taken after the antibiotic
therapy has ended. The safest place to take a blood sample is the
dorsal side of the newborn's heel. The marked circle must be
completely filled.
with blood, it does not matter if the blood has crossed the edges of
the circle. Before injecting the child, you should wait until the
disinfectant used to wipe the skin is completely dry. Otherwise, a
disinfectant is mixed with the blood sample, and such a sample is
unusable. Iodine and means containing iodine are not used because
they interfere with the determination of thyrotropin for diagnosing
congenital hypothyroidism. It is important to write on the back of
the paper whether the child is taking antibiotics and is seriously
ill.
Screening for congenital hypothyroidism
Congenital hypothyroidism can be diagnosed late or go completely
undiagnosed, causing health disorders for the child, economic and
social burden for the family. Therapeutic treatment of diagnosed
congenital hypothyroidism is simple, cheap and effective. With early
diagnosis and therapy, the newborn develops normally without mental
handicap and becomes a productive member of society. The child's
suffering, the economic and social burden caused by congenital
hypothyroidism, obliged the institutions of many countries to
include newborn screening for hypothyroidism as a mandatory form of
child health care.
In Montenegro, screening for hypothyroidism was introduced as a
mandatory form of child health care in 2008. To date, congenital
hypothyroidism is the only endocrine disease included in the newborn
screening program.
The main clinical features of untreated congenital hypothyroidism
are growth failure and delayed neurocognitive development resulting
in mental retardation.
Figure 2. Clinical picture of congenital
hypothyroidism
https://www.researchgate.net/publication/44662677/figure/fig4/AS:279090520182836@1443551773718/
Infant-with-congenital-hypothyroidism-A-3-month-old-infant-with-untreated-CH-picture_Q320.jpg
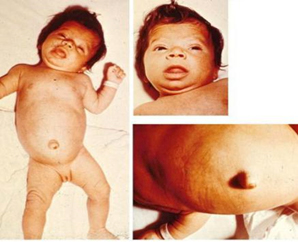
Worldwide, the incidence rate of congenital hypothyroidism is 1:
2000-4000 newborns, while areas that are deficient in iodine record
a higher incidence rate [10]. Congenital hypothyroidism is diagnosed
at birth using the Guthrie test. This test is based on measuring the
value of TSH or T4 (thyroxine). If the level of T4 in the blood from
the heel prick is low and the TSH is elevated, the screening results
indicate the development of congenital hypothyroidism. Confirmation
of the diagnosis is made by analyzing hormones from venous blood,
where the level of TSH and T4 is also measured. If the value of T4
hormone is low, and the value of TSH is elevated, the diagnosis is
confirmed [11].
The goal of hormone replacement therapy is to bring the child to a
state of euthyroidism. In diagnosed congenital hypothyroidism,
therapy is started with a full dose of hormones in order to prevent
or reduce the harmful effects of hypothyroidism on the development
of the central nervous system. It is recommended to maintain the
concentration of T3 and T4 at the upper limit of normal. At the
beginning of the therapy, the level of T4 and T3 is normalized and
the elevated TSH is suppressed. With well-managed therapy, normal
growth is achieved and clinical signs of hypothyroidism disappear,
but the prognosis of mental development is not so favorable and
depends above all on the time when the therapy was started.
Levothyroxine is a hormonal preparation that is used in the form of
tablets or solutions. The tablet should be crushed and mixed with 30
ml of liquid (water, milk or formula). The solution is given to the
child through a syringe or pipette, it should not be mixed with the
entire meal in the bottle because it may happen that the baby does
not eat the entire meal and the full dose of the medicine is not
taken. During hormone therapy, it is necessary to monitor the
condition of the child, because due to an overdose with
levothyroxine, symptoms of hyperthyroidism may develop:
restlessness, mild diarrhea, slow progress in body weight, insomnia,
accelerated growth.
Due to an insufficient therapeutic dose, the child may develop
lethargy, constipation, cold extremities, unexpected weight gain,
and slow growth.
After starting hormone therapy, it is necessary to monitor the
values of thyroid hormones. In the first months, the hormonal status
is checked every few weeks, ie every three to six months during
childhood, or every 6 to 12 months in adulthood [12]. A large number
of countries have included hypothyroidism in their newborn screening
program, in such a way that from the same filter paper blood sample
that is taken to look for phenylketonuria, T4 or TSH is determined
radioimmunological.
Newborn screening for galactosemia
Due to lack of galactose-1-phospho-uridyl-transferase, classic
galactosemia occurs [13]. Due to the inactivity of this transferase,
galactose-1-phosphate accumulates in the liver, erythrocytes,
spleen, eye lens, kidneys, heart muscle and cerebral cortex, and
there is galactosemia in the blood. Besides the intracellular
accumulation of galactose and galactose-1-phosphate, there is also a
larger amount of galactitol. After a few days of feeding with
mother's milk or milk formula containing lactose, the newborn
becomes anorexic and turns yellow. Infants with classic often refuse
food, do not progress or lose weight, vomit after meals, have
diarrhea, jaundice, ascites, edema, hepatomegaly, are lethargic and
hypotonic. Liver damage can progress to fulminant failure with
encephalopathy and hemorrhagic diathesis, and renal failure is
possible [14].
Figure 3. A child with galactosemia
https://encrypted-tbn0.gstatic.com/images?q=tbn:ANd9GcTpVTHhntyHltIfN9_
IwAGV4X8QUKZkDzQ51mKrGQqKsz5XitFfyvnvkKHrwiQSg4ZNKxA&usqp=CAU
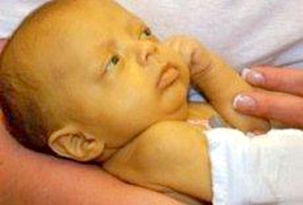
Children remain short with speech defects as well as posture and
balance disorders during adolescence. Accumulation of galactose and
galactitol in the eye lens leads to the rapid formation of
cataracts, clouding of the eye lens and loss of vision. The disease
can be accompanied by osteomalacia, temporary ovarian failure, while
more severe forms of galactosemia are accompanied by hearing loss
[15]. The treatment of galactosemia is based on a diet without any
galactose (for infants it is soy milk instead of cow's milk). It
should be started at the first suspicion of this disease, without
waiting for the test results. If the diet is started in time, the
symptoms can gradually disappear. The long-term prognosis of treated
children is good, although some of them may have a slight delay in
growth, mild speech difficulties and other discrete mental
disorders. Patients have elevated concentrations of galactose in
serum and urine. A woman who knows she carries the gene for
galactosemia must also completely stop eating foods containing
galactose during pregnancy. Galactosemia can be prevented during
pregnancy with an appropriate diet. If the mother has a high level
of galactose in her blood, it can pass through the placenta and
cause cataracts. People with this disorder must give up galactose
for life [16].
Screening for glutaric aciduria type I
Glutaric aciduria type 1 is a severe inherited neurometabolic
disorder whose clinical outcome has improved after the
implementation of a newborn screening program and prompt initiation
of presymptomatic metabolic treatment.
Glutaric acidemia type I is the antitype of the so-called cerebral
organic aciduria and is the result of a hereditary disorder in the
metabolism of the amino acids lysine, hydroxylysine and tryptophan,
due to the lack of the mitochondrial enzyme
glutaryl-CoA-dehydrogenase. In patients with enzyme deficiency,
glutaric and, to a lesser extent, 3-OH-glutaric and glutaconic acid
accumulate in the brain [17]. The estimated prevalence of the
disease ranges from 1:125,000 to 1:250 newborns in genetically
high-risk populations [18]. Untreated disease most often causes a
picture of acute brain damage with severe dystonic-dyskinetic
disorder (Figure 6). The disease is asymptomatic until the age of
usually half a year to a year, when the child develops the
so-called. encephalopathic crisis in which the basal ganglia are
affected.
Figure 4. Child with glutaric aciduria type
https://upload.wikimedia.org/wikipedia/commons/thumb/1/19/GA1_
posture2.jpg/220px-GA1_posture2.jpg

The disease is characterized by neurodevelopmental disorders,
including: delay/deficit in speech development, learning
difficulties, intellectual development disorder, epilepsy,
macrocephaly [19]. Combined metabolic therapy includes a low-lysine
diet, carnitine supplementation, and emergency treatment during the
episode to prevent catabolism and minimize CNS exposure to lysine
and its toxic metabolic byproducts [20].
Screening for cystic fibrosis
Neonatal screening for cystic fibrosis has optimized patient
prognosis by enabling very early multidisciplinary care. Over the
past 20 years, screening programs have experienced a major
international expansion. Cystic fibrosis is included in the
screening program in Serbia. In the middle of the 20th century, when
the disease was discovered, children suffering from cystic fibrosis
died within the first year of life. With early diagnosis, improved
treatment and the use of new drugs, the average life expectancy of
sufferers is 40 years. In countries that have introduced neonatal
screening, the life expectancy of patients has been significantly
extended, and the quality of life of patients and their families has
improved.
Cystic fibrosis is an autosomal recessive disease characterized by
pancreatic insufficiency and chronic endobronchial infection of the
respiratory tract. Chronic airway infection leads to progressive
bronchiectasis and ultimately respiratory failure, which is the
leading cause of death in patients with cystic fibrosis. Other
complications include sinusitis, diabetes mellitus, intestinal
obstruction, hepatobiliary disease, hyponatremic dehydration, and
infertility [21].
The advantage of early diagnosis of cystic fibrosis through neonatal
screening is multiple: application of preventive and early
therapeutic interventions, regular control and early detection of
complications, significantly better survival of patients, longer and
better quality of life of patients, slower progression of lung
disease, prevention of malnutrition, better nutrition, normal growth
and child development.
CONCLUSION
Detection of the disease at the earliest age enables a quick
therapeutic approach, thus ensuring adequate psychophysical growth
and development of the child and preventing permanent physical and
intellectual deficits. Hereditary metabolic and endocrinological
diseases are characterized by a high percentage of physical and
mental disability, which affects not only the health and social
functioning of the child, but it affects the whole family, community
and society. Screening for congenital hypothyroidism began in
Montenegro in 2007. It is the only endocrinological hereditary
disorder that is included in the screening program in Montenegro.
From the surrounding countries Croatia has the largest number of
diseases included in the screening program, eight diseases:
phenylketonuria, hypothyroidism, three fatty acid breakdown
disorders, glutaric aciduria type 1, isovaleric aciduria, carnitine
carrier deficiency.
REFERENCES
- Advisory Committe on Heritable Disorders in Newborn and
children; Recommended Uniform Screening Panel. [Internet] [Citirano
2021 Novembar 02]. Dostupno na: https://ww.hrsa.gov/advisory-committes/heritable
dosorders/rusp/index.html
- Stone WL, Basit H, Los E. Phenylketonuria. 2021 Nov 5. In:
StatPearls [Internet]. Treasure Island (FL): StatPearls
Publishing; 2022. [Citirano 2022 Avgust 01]. Dostupno na:
https://pubmed.ncbi.nlm.nih.gov/30570999/
- Woolf LI, Adams J. The Early History of PKU. Int J Neonatal
Screen. 2020;6(3):59. [Citirano 2022 Jul 28]. Dostupno na:
https://pubmed.ncbi.nlm.nih.gov/33239585/
- Mancilla VJ, Mann AE, Zhang Y, Allen MS. The Adult
Phenylketonuria (PKU) Gut Microbiome. Microorganisms.
2021;9(3):530. [Citirano 2022 Avgust 04]. Dostupno na:
https://pubmed.ncbi.nlm.nih.gov/33806544/
- Wiedemann A, Oussalah A, Jeannesson É, Guéant JL, Feillet F.
La phénylcétonurie - De la diététique à la thérapie génique [Phenylketonuria,
from diet to gene therapy]. Med Sci (Paris).
2020;36(8-9):725-734. [Citirano 2022 Avgust 04]. Dostupno na:
https://pubmed.ncbi.nlm.nih.gov/32821049/
- van Spronsen FJ, Blau N, Harding C, Burlina A, Longo N,
Bosch AM. Phenylketonuria. Nat Rev Dis Primers. 202;7(1):36. [Citirano
2022 Avgust 04]. Dostupno na:
https://pubmed.ncbi.nlm.nih.gov/34017006/
- Chen S, Zhu M, Hao Y, Feng J, Zhang Y. Effect of Delayed
Diagnosis of Phenylketonuria With Imaging Findings of Bilateral
Diffuse Symmetric White Matter Lesions: A Case Report and
Literature Review. Front Neurol. 2019;10:1040. [Citirano 2022
Avgust 04]. Dostupno na:
https://pubmed.ncbi.nlm.nih.gov/31636599/
- MacDonald A, van Wegberg AMJ, Ahring K, Beblo S, Bélanger-Quintana
A, Burlinaet et al., APKU dietary handbook to accompany PKU
guidelines. Orphanet J Rare Dis. 2020;15(1):171. [Citirano 2022
Avgust 05]. Dostupno na:
https://pubmed.ncbi.nlm.nih.gov/32605583/
- Arduini GAO, Balarin MAS, Silva-Grecco RLD, Marqui ABT.
KNOWLEDGE OF PUERPERAL MOTHERS ABOUT THE GUTHRIE TEST. Rev Paul
Pediatr. 2017;35(2):151-157. [Citirano 2022 Avgust 05]. Dostupno
na: https://pubmed.ncbi.nlm.nih.gov/28977324/
- Guerri G, Bressan S, Sartori M, Costantini A, Benedetti S,
Agostini F et al., Hypothyroidism and hyperthyroidism. Acta
Biomed. 2019;90(10-S):83-86. [Citirano 2022 Avgust 05]. Dostupno
na: https://pubmed.ncbi.nlm.nih.gov/31577260/
- American thyroid association (2020). A review of the 2020
guidlines for congenital hypothyroidism. [Citirano 2021 Novembar
03] Dostupno na: thyroid.org/congenital-hypothyroidism
- British Thyroid foundation (2018). Congenital
hypothyroidism. [Citirano 2021 Novembar 02] Dostupno na :www.british-thyroid-association.org
- Yuzyuk T, Balakrishnan B, Schwarz EL, De Biase I, Hobert J,
Longo N et al., Effect of genotype on galactose-1-phosphate in
classic galactosemia patients. Mol Genet Metab. 2018
Nov;125(3):258-265. [Citirano 2022 Avgust 05]. Dostupno na:
https://pubmed.ncbi.nlm.nih.gov/30172461/
- Lak R, Yazdizadeh B, Davari M, Nouhi M, Kelishadi R. Newborn
screening for galactosaemia. Cochrane Database Syst Rev.
2017;12(12):CD012272. [Citirano 2022 Avgust 05]. Dostupno na:
https://pubmed.ncbi.nlm.nih.gov/29274129/
- Hrvatski liječnički zbor u saradnji sa farmaceutskom tvrtkom
MSD (2014) MSD priručnik dijagnostike i terapije. [Citirano 2022
Januar 06]. Dostupno na:
http://www.msd-prirucnici.placebo.hr/msd-prirucnik/pedijatrija/nasljedne-metaboličke-bolesti/galaktozemija
- Kiss E, Balogh L, Reismann P. Klasszikus galactosaemia
dietetikai kezelési lehetőségei [Diet treatment of classical
galactosemia]. Orv Hetil. 2017;158(47):1864-1867. Hungarian. [Citirano
2022 Avgust 05]. Dostupno na:
https://pubmed.ncbi.nlm.nih.gov/29153024/
- Boy N, Mohr A, Garbade SF, Freisinger P, Heringer-Seifert J,
Seitz A et al., Subdural hematoma in glutaric aciduria type 1:
High excreters are prone to incidental SDH despite newborn
screening. J Inherit Metab Dis. 2021;44(6):1343-1352. [Citirano
2022 Avgust 05]. Dostupno na:
https://pubmed.ncbi.nlm.nih.gov/34515344/
- Boy N, Mengler K, Heringer-Seifert J, Hoffmann GF, Garbade
SF, Kölker S. Impact of newborn screening and quality of therapy
on the neurological outcome in glutaric aciduria type 1: a
meta-analysis. Genet Med. 2021;23(1):13-21. doi:
10.1038/s41436-020-00971-4. Epub 2020 Sep 28. PMID: 32981931;
PMCID: PMC7790745.
- Pokora P, Jezela-Stanek A, Różdżyńska-Świątkowska A,
Jurkiewicz E, Bogdańska A, Szymańska E, Rokicki D, Ciara E,
Rydzanicz M, Stawiński P, Płoski R, Tylki-Szymańska A. Mild
phenotype of glutaric aciduria type 1 in polish patients - novel
data from a group of 13 cases. Metab Brain Dis.
2019;34(2):641-649. doi: 10.1007/s11011-018-0357-5. Epub 2018
Dec 20. PMID: 30570710; PMCID: PMC6428789.
- Larson A, Goodman S. Glutaric Acidemia Type 1.. In: Adam MP,
Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Mirzaa
GM, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA):
University of Washington, Seattle; 2019;1993–2022. PMID:
31536184.
- Goetz D, Ren CL. Review of Cystic Fibrosis. Pediatr Ann.
2019;48(4):e154-e161. doi: 10.3928/19382359-20190327-01. PMID:
30986316.
|
|
|
|