| |
|
|
INTRODUCTION: NEUROENDOCRINE CONTROL AND PHYSIOLOGY
1. Hypothalamic–Pituitary–Gonadal (HPG) Axis
Puberty is the result of reactivation of the hypothalamic–pituitary–gonadal
(HPG) axis [1]. This complex process occurs through three key
phases:
Fetal activation:
The HPG axis becomes active between the 12th and 14th week of
gestation, but is suppressed toward the end of pregnancy by
placental hormones [1].
Mini-puberty:
A short-term reactivation of the axis occurs immediately after birth
due to the removal of placental inhibition. It lasts up to 6 months
in boys, while in girls estradiol levels may fluctuate up to 2–4
years of age, leading to transient breast enlargement [1,2].
True puberty:
Occurs when neuroendocrine mechanisms (primarily the kisspeptin
system and leptin) remove central nervous system (CNS) inhibition of
GnRH neurons. This triggers pulsatile secretion of gonadotropin-releasing
hormone (GnRH), which stimulates the pituitary gland to release
luteinizing hormone (LH) and follicle-stimulating hormone (FSH),
thereby initiating gonadal maturation [1,3–6].
The main components of this regulatory system and their functions
are summarized in Table 1.
Table 1. Components and Regulation of the HPG
Axis. Source: Adapted from Sharma L, Daley SF [1]

2. Key Terms and Physiological Processes
Understanding pubertal disorders requires a clear distinction
between two independent processes:
- Gonadarche:
Activation of the gonads under the influence of the HPG axis. In
girls, it leads to ovarian growth and breast development (via
estradiol), while in boys it leads to testicular enlargement and
spermatogenesis (via testosterone) [2,7].
- Adrenarche:
Increased production of adrenal androgens (DHEA and DHEA-S). It
occurs independently of the HPG axis, around 7–8 years of age, and
is responsible for the development of pubic hair (pubarche), acne,
and body odor.
Hormonal and physical changes in normal development
Physical changes of puberty result from sex steroid production by
the gonads, and the onset of gonadarche indicates the beginning of
puberty. Gonadarche is initiated by pulsatile secretion of
gonadotropin-releasing hormone (GnRH), which activates the HPG axis
[1–3].
Adrenarche (i.e., adrenal androgen production leading to pubic and
axillary hair, body odor, and mild acne) is a separate but usually
concurrent process and, by itself, does not indicate true pubertal
onset in either boys or girls [8].
In girls, increased ovarian estradiol secretion leads to breast
development at an average age of 10 years (range: 8–12 years).
Menarche typically follows approximately 2.5 years after the onset
of breast development, at an average age of 12.5 years (range: 9–15
years) [1,2,3,7,9].
In boys, testicular enlargement to at least 4 mL in volume or 2.5 cm
in length is the first sign of true puberty and occurs at an average
age of 11.5 years (range: 9.5–14 years) [8,10].
Peak height velocity (PHV) occurs earlier in puberty in girls and
later in boys, with an average sex difference of approximately two
years [11].
At the onset of menarche, approximately 95.3% (SD 1.7) of adult
height has already been achieved; the remaining height gain averages
7.8 cm (SD 2.8) [12].
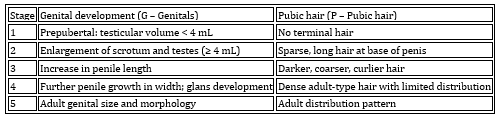
3. Clinical Progression (Tanner Stages)
Pubertal progression follows a predictable sequence of physiological
changes that are clinically assessed using the standardized Tanner
staging system (I–V) [1,13].
Detailed criteria for assessing breast development and pubic hair in
girls are systematized in Table 2, while parameters for evaluating
genital development and pubic hair in boys are presented in Table 3.
Table 2. Tanner Classification of Development in
Girls
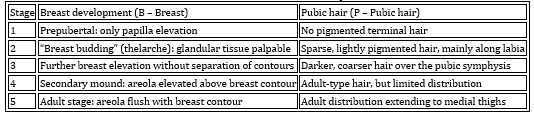
Table 3. Tanner Classification of Development in
Boys
ETIOLOGY AND CLASSIFICATION
Precocious puberty is defined as the appearance of secondary
sexual characteristics before the age of 9 years in boys (or before
8 years in girls), corresponding to a chronological age
approximately 2–2.5 standard deviations earlier than the average age
of pubertal onset in the White population [13,14]. Its incidence
ranges between 1:5,000 and 1:10,000, while its prevalence is
increasing worldwide [15].
Based on the underlying pathological mechanism, precocious puberty
can be classified as follows:
Central Precocious Puberty (CPP)
(Gonadotropin-dependent) – caused by early maturation of the HPG
axis. It results from premature activation of the axis (GnRH-dependent)
[16,2]. Etiologies include congenital abnormalities (hamartoma,
cysts), acquired lesions (tumors, trauma), and genetic mutations
(e.g., MKRN3). In girls, up to 90% of cases are idiopathic [2,9].
Peripheral Precocious Puberty (PPP)
(Gonadotropin-independent) – caused by excessive secretion of sex
steroids from the gonads or adrenal glands, exogenous exposure to
sex steroids, or ectopic production of gonadotropins from germ cell
tumors.
Benign Pubertal Variants
These include non-progressive or intermittently progressive forms of
CPP, as well as isolated androgen-mediated sexual characteristics in
boys resulting from early activation of the
hypothalamic–pituitary–adrenal axis (premature adrenarche). Both
conditions may represent normal variants of pubertal development
[13,14].
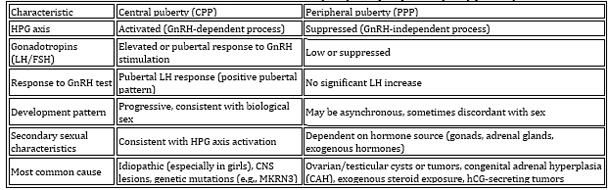
Differential characteristics between central and peripheral
precocious puberty are summarized in Table 4.
Table 4. Differential diagnosis of central (CPP)
and peripheral (PPP) puberty
Detailed description of peripheral precocious puberty (PPP):
Peripheral precocious puberty (PPP) is caused by excessive
production of sex steroids from the gonads or adrenal glands,
secretion of β-hCG–producing tumors, or exposure to exogenous sex
hormones. Etiological causes include McCune–Albright syndrome (MAS),
functional ovarian cysts (FC), Leydig cell tumors, or familial
male-limited precocious puberty. Adrenal sources of androgen excess
are most commonly due to adrenal tumors or congenital adrenal
hyperplasia [17]. PPP is significantly less common than central
precocious puberty (CPP).
Non-classic congenital adrenal hyperplasia (NCAH), most commonly due
to 21-hydroxylase deficiency (CYP21A2 gene mutation), is an
autosomal recessive disorder. Clinical manifestations reflect
androgen excess, including premature pubic hair (pubarche), body
odor, and acne before the age of 8 in girls or 9 in boys. Additional
features may include accelerated linear growth during childhood and
advanced bone maturation, which can ultimately result in reduced
adult height due to premature epiphyseal closure [18,19,20].
MFor accurate diagnosis of NCAH, assessment of
17-hydroxyprogesterone (17-OHP) levels—often including basal and
ACTH-stimulated values—is essential, as they correlate with disease
severity and are used for diagnostic confirmation (see Table 5)
Table 5. Differential diagnosis of NCCAH based on
17-OHP levels. Source: Adapted from White PC, Speiser PW [21]

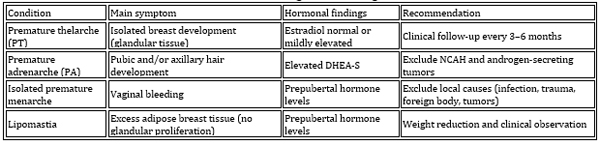
BENIGN VARIANTS (PARTIAL PRECOCIOUS PUBERTY)
Benign variants of precocious puberty include premature thelarche,
premature adrenarche, and isolated premature menarche. These
conditions are characterized by the appearance of isolated pubertal
signs without full activation of the hypothalamic–pituitary–gonadal
(HPG) axis. Importantly, bone age, growth velocity, and biochemical
findings are usually within normal limits [1,8]. Sharma L and Daley
SF emphasize the importance of distinguishing these conditions to
reduce unnecessary diagnostic procedures [1].
Premature thelarche (PT)
The most common benign variant. It presents as unilateral or
bilateral breast development in girls, typically occurring between
0–24 months of age or again around 6–8 years. No other pubertal
changes are present. Clinical follow-up is recommended to monitor
for progression to central puberty [1,22,23,24].
Premature adrenarche (PA)
Characterized by early adrenal androgen production, leading to pubic
or axillary hair, acne, and body odor before the age of 8 years.
There is no breast development or testicular enlargement. Exogenous
androgen exposure, tumors, and late-onset congenital adrenal
hyperplasia (CAH) must be excluded [1,24].
Isolated premature menarche
Defined as vaginal bleeding in girls younger than 8 years in the
absence of other pubertal signs. It generally does not affect final
adult height. Differential diagnosis must exclude sexual abuse,
foreign bodies, genital tract tumors, and infections [1,24].
Table 6. Differential diagnosis of benign variants
CLINICAL ASSESSMENT AND DIAGNOSTIC APPROACH
1. Medical history and anthropometry
A detailed clinical history is essential to distinguish true
precocious puberty (PP) from benign variants. Progressive pubertal
development, rapid linear growth, and advanced bone age are
characteristic of true PP [1,25].
The evaluation should include:
Neurological symptoms (headache, seizures, episodes of inappropriate
laughter – suggestive of hypothalamic hamartoma)
Previous head trauma, brain tumor treatment, or central nervous
system (CNS) infections
Physical examination: assessment of pubic and axillary hair, signs
of virilization (clitoromegaly, penile enlargement, acne), and full
neurological examination
Skin examination: café-au-lait macules (suggestive of
Neurofibromatosis type 1 or McCune–Albright syndrome)
Growth velocity: a growth spurt >7 cm/year with breast or testicular
enlargement requires urgent evaluation [24]
2. Laboratory and radiological evaluation
Bone age (BA):
Advanced bone age >2 standard deviations (SD) compared to
chronological age (CA) requires further diagnostic work-up [1,14].
Hormonal testing:
Measured using ultrasensitive assays (ICMA or ECLIA). Basal serum LH
levels >0.2–0.3 IU/L may indicate pubertal activation [1].
GnRH stimulation test (gold standard):
Activation of the pubertal HPG axis is confirmed if peak LH >5 IU/L.
An LH/FSH ratio <0.43 suggests a prepubertal state, while a
stimulated ratio >0.66 helps differentiate progressive from
non-progressive variants [1].
In girls:
Serum estradiol (E2) levels after 24-hour GnRH agonist stimulation
(peak >50 pg/mL) improve diagnostic sensitivity [16,22].
In boys:
Measurement of testosterone, DHEA-S, 17-OHP, and early-morning hCG
is recommended when PPP is suspected. Certain tumors may secrete hCG,
which activates LH receptors and mimics central puberty [1].
Reference tables:
Reference hormone and steroid levels are presented in Table 7 and
Table 8
Pelvic ultrasound criteria in girls are shown in Table 8
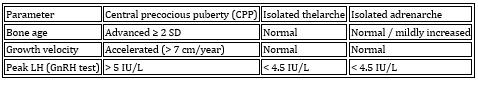
Differential diagnostic criteria (CPP vs benign variants) are
presented in Table 9.
Table 7. Reference serum concentrations of
gonadotropins and steroids. Source: Neely EK et al. [26]

Table 8. Pelvic ultrasound criteria in girls
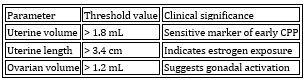
Table 9. Differential diagnostic criteria (CPP vs
benign variants)
DIAGNOSTIC ALGORITHMS
ALGORITHM 1. DIAGNOSTIC APPROACH IN GIRLS WITH THELARCHE
(Adapted from: Root AW. Pediatr Rev. 2000 [27])
Normal growth velocity and bone age (BA ≈ CA):
Bone age corresponds to chronological age.
Diagnosis: Isolated premature thelarche
Management: Clinical follow-up; no treatment usually required
Accelerated growth velocity and advanced bone age (BA > CA):
Bone age is advanced compared to chronological age.
Indicated test: GnRH stimulation test
Peak LH > 5 IU/L (pubertal response):
Diagnosis: Central precocious puberty (CPP)
Next step: Brain MRI to exclude CNS pathology
Low LH (prepubertal response) with ovarian cysts present:
Suspicion: McCune–Albright syndrome (MAS) or other forms of
peripheral puberty
In boys, differential diagnosis requires a systematic approach
presented in Algorithm 2.
ALGORITHM 2. DIAGNOSTIC EVALUATION OF BOYS WITH PRECOCIOUS
PUBERTY (Adapted from: Root AW. Pediatr Rev. 2000 [27])
I. Clinical triage (growth velocity and bone age assessment)
BA ≈ CA:
Likely isolated premature adrenarche → Periodic clinical follow-up
recommended
BA > CA: → Requires hormonal laboratory evaluation
II. Gonadotropin (LH) assessment
Elevated LH (pubertal response at baseline or after GnRH test):
Diagnosis: Central precocious puberty (CPP)
Mandatory: Brain MRI to exclude hypothalamic hamartoma or CNS tumors
Low LH (suppressed/prepubertal response):
Diagnosis: Peripheral precocious puberty
→ Proceed with etiological work-up
III. Differential diagnosis of peripheral precocious puberty
(low LH)
Elevated 17-OHP / DHEA-S:
Suggests congenital adrenal hyperplasia (CAH) or adrenal tumors
Elevated hCG:
Suggests ectopic hCG-secreting tumors (e.g., hepatoblastoma or germ
cell tumors)
High testosterone with suppressed gonadotropins and enlarged testes:
Suggests testotoxicosis (familial male-limited precocious puberty,
FMPP) or Leydig cell tumor
THERAPY AND MANAGEMENT
1. Central precocious puberty (CPP)
Gold standard treatment: GnRH agonists (GnRHa) [7,24]
Goals: Maximize final adult height and reduce psychosocial stress
Early onset (<6–7 years) with rapid progression → standard
indication for treatment
Formulations:
Monthly depot injections (3.75 mg)
Long-acting depot preparations (every 4–12 weeks)
Monitoring:
Clinical evaluation every 3–6 months
Bone age every 6–12 months
Target stimulated LH suppression: <2.5–4.5 IU/L
Discontinuation:
Usually around chronological age 11 years
Or when bone age reaches ~12.5 years in girls and ~14 years in boys
[1,7,28]
Safety:
Therapy is considered safe
Meta-analysis shows average gain in final height of ~0.63 SDS [1]
2. Peripheral precocious puberty (PPP)
Surgery: For gonadal or adrenal tumors
NCCAH: Treated with glucocorticoids
MAS: Aromatase inhibitors and selective estrogen receptor modulators
Important note: Children with PPP may later develop secondary
CPP; in such cases, GnRH analogs should be added [1]
CONCLUSION (Practical aspects)
The main clinical sign suggesting precocious puberty is the
development of breast tissue in girls and testicular enlargement (>
4 mL) in boys before 8–9 years of age.
Differential diagnosis: The priority is to distinguish benign
variants from progressive central precocious puberty (CPP) in order
to avoid unnecessary treatment.
Gold standard: The GnRH stimulation test combined with assessment of
bone age maturation.
Brain MRI: Recommended in all cases of CPP in boys, and in girls
younger than 6 years or in those with neurological symptoms.
Time is a critical factor: The best outcomes are achieved when
treatment is initiated before 6 years of age.
Education: A thorough discussion with the family is
essential, including explanation of normal pubertal development,
treatment goals, and psychosocial aspects (peer interaction,
self-esteem, and emotional well-being).
LITERATURE:
1. Sharma L, Daley SF. Precocious Puberty. [Updated 2025 Nov 7].
In: StatPearls [Internet].
2. Cheuiche AV, et al. Diagnosis and management of precocious sexual
maturation. Eur J Pediatr. 2021.
3. Alghamdi A. Precocious Puberty: Types, Pathogenesis and Updated
Management. Cureus. 2023.
4. Largo RH, Prader A. Somatische Pubertätsentwicklung bei Mädchen.
Monatsschr Kinderheilkd. 1987.
5. Marshall WA, Tanner JM. Variations in pattern of pubertal changes
in girls. Arch Dis Child. 1969.
6. Marshall WA, Tanner JM. Variations in the pattern of pubertal
changes in boys. Arch Dis Child. 1970.
7. Bonomi M, et al. Management of andrological disorders. J
Endocrinol Invest. 2025.
8. Klein DA, et al. Disorders of Puberty: An Approach to Diagnosis
and Management. Am Fam Physician. 2017.
9. Sizonenko PC. Normal sexual maturation. Pediatrician. 1987.
10. Kang E, et al. Etiology and therapeutic outcomes of children
with PPP. Ann Pediatr Endocrinol Metab. 2016.
11. Luo X, et al. Long-term efficacy and safety of GnRHa treatment.
Clin Endocrinol. 2021.
12. Baek JW, et al. Age of menarche and near adult height after
long-term GnRHa treatment. Ann Pediatr Endocrinol Metab. 2014.
13. Wheeler MD. Physical changes of puberty. Endocrinol Metab Clin
North Am. 1991.
14. Taranger J, et al. VI. Somatic pubertal development. Acta
Paediatr Scand Suppl. 1976.
15. Beştaş A, et al. Evaluation of Clinical and Laboratory Findings.
Indian J Endocrinol Metab. 2023.
16. Bangalore Krishna K, Garibaldi L. Critical appraisal of
diagnostic laboratory tests. Front Pediatr. 2025.
17. Cavarzere P, et al. Revising LH cut-off for the diagnosis of CPP.
Endocrine. 2025.
18. Witchel SF, Azziz R. Nonclassic congenital adrenal hyperplasia.
Int J Pediatr Endocrinol. 2010.
19. Witchel SF. Non-classic congenital adrenal hyperplasia.
Steroids. 2013.
20. Loli P, et al. Non-classical congenital adrenal hyperplasia:
current insights. Endocrine. 2025.
21. White PC, Speiser PW. Congenital adrenal hyperplasia. Endocr
Rev. 2000.
22. Cappa M, Chioma L. Disorders of Pubertal Development. Springer;
2021.
23. Della Manna T, et al. Premature thelarche: identification of
clinical and laboratory data. Rev Hosp Clin. 2002.
24. Paparella R, et al. Precocious Puberty and Benign Variants in
Female Children. Endocrines. 2025.
25. Widek T, et al. Bone age estimation with the Greulich-Pyle atlas
using 3T MR images. Forensic Sci Int. 2021.
26. Neely EK, et al. Normal ranges for immunochemiluminometric
gonadotropin assay. J Pediatr. 1995.
27. Root AW. Precocious puberty. Pediatr Rev. 2000.
28. Kilberg MJ, Vogiatzi MG. Approach to the Patient: Central
Precocious Puberty. J Clin Endocrinol Metab. 2023.
ABBREVIATIONS:
ACTH – Adrenocorticotropic hormone
BMI – Body mass index
CNS – Central nervous system
CPP – Central precocious puberty
DHEA-S – Dehydroepiandrosterone sulfate
FSH – Follicle-stimulating hormone
GnRH – Gonadotropin-releasing hormone
GnRHa – Gonadotropin-releasing hormone agonists
HPG axis – Hypothalamic–pituitary–gonadal axis
CAH – Congenital adrenal hyperplasia
LH – Luteinizing hormone
MAS – McCune–Albright syndrome
MRI – Magnetic resonance imaging
NCAH – Non-classic congenital adrenal hyperplasia
PPP – Peripheral precocious puberty
SDS – Standard deviation score
TSH – Thyroid-stimulating hormone
BA – Bone age
CA – Chronological age
US – Ultrasound
|
|
|
|