| |
|
|
UVOD
Povezanost između hronične bubrežne bolesti (HBB) i
kardiovaskularnih (KV) abnormalnosti, prvi put je zabeležio dr
Richard Bright još 1836. godine. On je zapazio da se strukturne
promene u srcu bubrežnih bolesnika sastoje uglavnom od hipertrofije
čiji je stepen proporcionalan dužini trajanja bubrežne bolesti, pa
je pretpostavio da promenjeni kvalitet krvi, naročito u perifernoj
vaskulaturi, zahteva povećanu silu koja bi državala cirkulaciju.
Uzimajući u obzir da je bubrežna bolest primarni poremećaj, a KV
promene su sekundarne, Bright je postavio koncept bubrežnog porekla
KV bolesti (KVB) [1]. Mnoge epidemiološke studije od tada potvrdile
su ovu povezanost, ali su pružile tek delimične odgovore na brojna
pitanja, pre svega zbog činjenice da klinička istraživanja u oblasti
kardiologije, po pravilu, isključuju bolesnike sa značajnom HBB [2].
Sa druge strane, patofiziološka istraživanja ukazuju da postupci
usmereni na prevenciju progresije HBB takođe preveniraju i KV
morbiditet i mortalitet.
Kardiovaskularni rizik i kardiovaskularno oboljevanje u HBB
Pionirski radovi Lindnera i saradnika iz 1974. godine pokazali su da
bolesnici lečeni ponavljanim hemodijalizama imaju ubrzani razvoj
ateroskleroze i da umiru od KVB u ranijem životnom dobu nego opšta
populacija [3]. Inicijalno se smatralo da je povećani KV rizik
ograničen samo na ovu populaciju bolesnika sa terminalnom HBB, pa je
izračunato da je, nakon stratifikacije prema starosti, polu, rasi i
prisustvu ili odsustvu diabetes mellitus-a, KV mortalitet dijaliznih
bolesnika 10-20 puta veći nego u opštoj populaciji [4]. Međutim,
kasnije studije pokazale su da se povećani KV rizik prostire kroz
čitav spektar i sve stadijume HBB, pa su već povećana albuminurija i
blago snižena jačina glomerulske filtracije (JGF) udružene sa
povećanom učestalošću KVB [5, 6]. Istraživanje apsolutnog KV rizika
u velikoj kohorti opšte populacije u Kanadi pokazalo je da
tridesetogodišnji bolesnici sa HBB u stadijumu 3B imaju skraćenje
očekivane dužine života za 17 godina, a u stadijumu 4 za 25 godina u
poređenju sa osobama koje nemaju HBB, što znači da je očekivana
dužina života značajno redukovana kod bolesnika sa oštećenom
bubrežnom funkcijom [7].
Kliničke studije su pokazale da se kod bolesnika sa blagom do
umerenom HBB (stadijumi 3A i 3B) registruje mnogo viša incidenca KV
mortaliteta nego incidenca bubrežne insuficijencije, što ukazuje da
su ove osobe u većoj meri opterećene povećanim rizikom KVB nego
rizikom terminalnog stadijuma HBB koji zahteva terapiju zamene
renalne funkcije [5, 6, 8]. Povećani KV rizik u HBB uslovljen je
delovanjem brojnih tradicionalnih, ali i pojavom netradicionalnih,
specifičnih faktora rizika koji se razvijaju tokom progresije HBB, a
koji udruženi, bolesnike sa HBB svrstavaju među najrizičnije grupe
za razvoj KVB. Spektar KVB u HBB uključuje ishemijsku (koronarnu)
bolest srca, kongestivnu srčanu insuficijenciju, aritmije i
perifernu vaskularnu bolest.
Faktori kardiovaskularnog rizika u hroničnoj bubrežnoj bolesti
S obzirom da je HBB često rezultat hipertenzije i diabetes
mellitus-a, povećani kardiovaskularni rizik kod bubrežnih bolesnika
se dugo smatrao posledicom ovih osnovnih bolesti. Međutim, meta
analize su jasno pokazale da su oštećena bubrežna funkcija i
povećana albuminurija faktori kardiovaskularnog rizika potpuno
nezavisni od prisustva hipertenzije ili diabetes mellitus-a [9, 10].
Nedavno je dokazano da, u populaciji bolesnika sa HBB, procena JGF i
albuminurije poboljšava predikciju KV rizika, pri čemu je
poboljšanje veće za KV mortalitet i srčanu insuficijenciju, nego za
koronarnu arterijsku bolest i moždani udar [11]. Ovo potvrđuje da je
povezanost bubrežne funkcije i albuminurije sa KV rizikom nezavisna
od tradicionalnih faktora KV rizika i da netradicionalni, za bubreg
specifični mehanizmi, mogu značajno doprineti KV riziku.
Razjašnjenje ovih mehanizama bi moglo pomoći smanjenju KV rizika i
poboljšanju ishoda kod osoba sa HBB [12].
Tradicionalni faktori rizika za KVB su istovremeno i faktori rizika
za HBB, te su, stoga, sa visokom učestalošću prisutni u populaciji
bolesnika sa HBB. Osim hipertenzije i diabetes mellitus-a, oni
obuhvataju i stariju životnu dob, dislipidemiju, pušenje i
gojaznost. Netradicionalni ili „novi“ faktori rizika su specifični
za uremiju i, samim tim, mnogo češći kod bolesnika sa HBB nego u
opštoj populaciji. Oni uključuju albuminuriju, anemiju,
hiperparatireoidizam, metaboličku bolest kostiju,
hiperhomocistinemiju, malnutriciju, izoforme apolipoproteina,
inflamaciju, endotelnu disfunkciju i oksidativni stres. Različiti
tradicionalni i netradicionalni faktori rizika pokazuju tendenciju
da deluju aditivno i ubrzavaju razvoj ateroskleroze i progresiju HBB
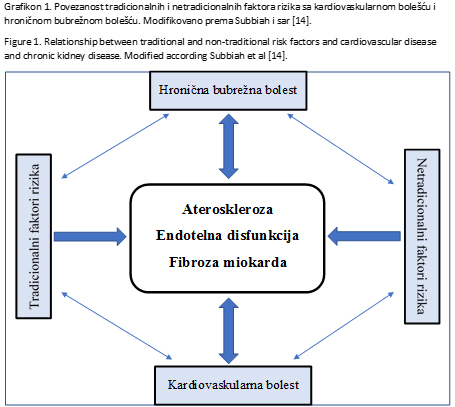
[2, 13]. Kompleksna povezanost između tradicionalnih i
netradicionalnih faktora rizika sa KVB i HBB je prikazana na
grafikonu broj 1 [14].
PATOFIZIOLOŠKI MEHANIZMI
Hipertenzija je dobro poznat i značajan faktor rizika za razvoj
HBB, ali i HBB, čak i u ranom stadijumu, može izazvati hipertenziju
koja je dalje odgovorna za povećanje KV rizika i ubrzanu progresiju
HBB [2]. Brojni su mehanizmi koji učestvuju u razvoju hipertenzije u
HBB, a oni utiču i na osnovne principe lečenja. Hipertenzija u HBB
se primarno razvija i održava usled disbalansa različitih
vazoaktivnih supstanci – aktivaciji/ nedovoljnoj supresiji
vazokonstriktornih sistema (renin-angiotenzin-aldosteron sistem
[RAAS], simpatički nervni sistem) i smanjenoj produkciji
vazodilatatornih agenasa (azot oksid [NO], prostaglandini).
Povećanje simpatičkog tonusa je posredovano aferentnim signalima
koji se generišu usled ishemije u funkcionalno oštećenim bubrezima.
Koncentracije kateholamina su, takođe, povišene u cirkulaciji
bolesnika sa HBB usled snižene aktivnosti renalaze, enzima koji se
stvara uglavnom u bubrezima i igra ulogu u inaktivaciji kateholamina
[2, 12, 15]. Sa opadanjem JGF dolazi do ushodne regulacije RAAS koji
podstiče retenciju vode i soli. Visok nivo angiotenzina II povećava
intracelularni sadržaj Na i Ca, izaziva vazokonstrikciju i povećanje
krvnog pritiska. Višak oba primarna medijatora RAAS-a, angiotenzina
i aldosterona, dovodi do povećanja endotelne rigidnosti u krvnim
sudovima i fibroze srca i bubrega, što direktno doprinosi razvoju
KVB i HBB [2, 12, 16]. Prekomerna stimulacija oba vazokonstriktorna
sistema stimuliše produkciju superoksida, interleukina 6 i drugih
proinflamatornih citokina. Sa druge strane smanjuje se dostupnost
NO, koji je uključen u kontrakciju i rast vaskularnih glatkih
mišićnih ćelija, agregaciju trombocita i adheziju leukocita za
endotel [12]. Drugi ključni faktor za endotelnu funkciju je
asimetrični dimetil arginin (ADMA), koji inhibira produkciju NO,
redukuje udarni volumen i povećava sistemsku vaskularnu rezistenciju
i krvni pritisak (KP) [17]. Koncentracija ADMA-a se povećava sa
opadanjem bubrežne funkcije i prognostički je značajna za mortalitet
i kardiovaskularne komplikacije kod bolesnika sa HBB [18]. Povećana
koncentracija ADMA-a zajedno sa simpatičkom hiperaktivnošću je
snažno povezana sa koncentričnom hipertrofijom leve komore (HLK)
[17].
Endotelna disfunkcija je karakteristika uznapredovale HBB (JGF <30
mL/min/1,73m2) i njena povezanost sa hipertenzijom je dobro
utemeljena [19]. Povećana rigidnost arterija se nalazi kroz čitav
spektar HBB, učestvuje u razvoju hipertenzije i nezavisan je faktor
rizika za KV događaje [20]. Od ostalih faktora, sekundarni
hiperparatireoidizam je udružen sa povećanjem intracelularne
koncentracije Ca i može voditi vazokonstrikciji i hipertenziji.
Hipertenzija se može pogoršati i usled terapije renalne anemije
rekombinantnim humanim eritropoetinom. U sistolnoj hipertenziji,
glavnu ulogu igraju kalcifikacije, smanjena elastičnost velikih
arterija, kao i često prisustvo hiperkinetičke cirkulacije
uzrokovane anemijom i kreiranjem arteriovenske fistule kao
vaskularnog pristupa za hemodijalizu. Kako se filtraciona funkcija
bubrega pogoršava i bubrežna slabost progredira, retencija vode i
soli preuzima esencijalnu ulogu u održavanju hipertenzije, vodeći
povećanju volumena plazme i udarnog volumena [21].
HLK je najčešći poremećaj srčane strukture i funkcije u HBB. U
populaciji bolesnika sa HBB koji imaju JGF nižu od 30 ml/min/1,73m2,
HLK se može detektovati kod 50% bolesnika, pri čemu je dominantna
koncentrična hipertrofija [22]. Prema istraživanjima Foley-a i
saradnika sprovedenim u grupi od 433 bolesnika sa terminalnim
stadijumom HBB, prevalenca HLK na početku dijaliznog lečenja je 74%
[23]. Poreklo HLK u HBB je multifaktorijalno. Patofiziološki faktori
obuhvataju opterećenje pritiskom, opterećenje volumenom i faktore
povezane sa uremijom. Pritom, hipertenzija i arterioskleroza
izazivaju opterećenje pritiskom i dovode do koncentrične HLK, dok
anemija, hipervolemija i arteriovenska fistula izazivaju opterećenje
volumenom i primarno dovode do dilatacije leve komore sa
ekcentričnom hipertrofijom. Na početku, HLK može biti kompenzatorni
proces koji omogućava levoj komori da poveća srčani rad i održi
konstantni nivo istegnutosti komorskog zida. Međutim, trajno
opterećenje odlikuje povećana brzina potrošnje energije, a nastali
disbalans između potrošnje i proizvodnje energije dovodi do
hroničnog energetskog deficita i smrti miocita. U uslovima relativno
smanjene kapilarne gustine i zadebljanja intramiokardnih arteriola,
napreduju maladaptivne strukturne promene u kojima dominira
akumulacija kolagena i intersticijalna fibroza miokarda [24].
Fibroza vodi progresivnom pogoršanju kontraktilnosti sa povećanjem
krutosti zida miokarda, sistolnoj i dijastolnoj disfunkciji,
dilatativnoj kardiomiopatiji i kongestivnoj srčanoj insuficijenciji.
U razvoju HLK kod bolesnika sa HBB učestvuju i nehemodinamski
faktori, kao što je povećani oksidativni stres i aktivacija ksantin
oksidaze. Aktivacija RAAS-a izaziva hiperaldosteronemiju koja
podstiče kardijalnu fibrozu putem produkcije profibrotičkog
transformišućeg faktora rasta. HLK može biti podstaknuta primenom Fe
ili eritropoetina u terapiji renalne anemije, ili deficitom vitamina
D. Novije studije ukazale su na nove biomarkere uključene u
patogenezu HLK. Jedan od njih je fibroblastni faktor rasta FGF23,
koji je primarno uključen u poremećaj metabolizma minerala i kostiju
i sekundarni hiperparatireoidizam. Povećani nivo FGF23 je najranije
detektovana serumska abnormalnost kod bubrežnih bolesnika sa
poremećenim metabolizmom minerala i kostiju, i njegov nivo postepeno
raste sa opadanjem bubrežne funkcije. Rezultati više kliničkih
studija su potvrdili postojanje uske povezanosti između FGF23 i HLK
[25]. Vitamin D je, takođe, uključen u regulaciju imunog,
kardiovaskularnog i endokrinog odgovora kroz aktivaciju nuklearnog
vitamin D receptora visokog afiniteta (VDR). Nedavno je ispitivana
povezanost između HLK i VDR genskog polimorfizma i pokazano je da je
VDR BsmI genski polimorfizam uključen u razvoj HLK u terminalnom
stadijumu HBB i nezavisno povezan sa progresijom HLK kako kod
dijaliznih, tako i kod predijaliznih bolesnika [26, 27]. Najnovija
istraživanja se fokusiraju na otkrivanje još specifičnijih
mehanizama kako bi se identifikovali novi terapijski ciljevi kod
bubrežnih bolesnika. Endogene mikro RNA (miRNA) su kratke,
ne-kodirajuće vrste RNA, koje su post-transkripcioni regulatori
usmereni na specifične mRNA, što rezultira u supresiji proteinske
sinteze ili u povećanju degradacije mRNA putem komplementarnog
vezivanja i na taj način utiče na ćelijsku funkciju. Poremećena
regulacija specifičnih miRNA se sreće kao ključni patološki faktor u
mnogim KVB. Tako, npr. miRNA-212/132 klaster je identifikovana kao
centralni regulator u razvoju HLK izazvane opterećenjem pritiskom i
srčane insuficijencije, putem represije antihipertrofičnog
transkripcionog faktora FOXO3[28]. Međutim, još uvek ne postoje
dokazi koji potvrđuju kardijalnu ekspresiju miRNA u HBB, što otvara
vrata za nova istraživanja.
Visoka prevalenca HLK sa pridruženim rizikom poremećaja srčanog
ritma može, bar delimično, objasniti zašto je prevalenca iznenadne
srčane smrti povećana u HBB. U opštoj populaciji, iznenadna srčana
smrt se pojavljuje sa učestalošću 1/1000 osoba-godina i smatra se
uzrokom 6-13% svih smrti, dok se među osobama sa HBB pojavljuje sa
stopom od 59/1000 osoba-godina i čini 26% svih uzroka smrti. Pored
visoke prevalence HLK, poremećen elektrolitni balans i povećana
prevalenca koronarne arterijske bolesti su predisponirajući faktori
za iznenadnu srčanu smrt u bolesnika sa HBB [29].
Valvularna srčana bolest i ateroskleroza se javljaju već u ranijim
stadijumima HBB, sa višom prevalencom nego u opštoj populaciji. Za
vaskularnu bolest kod bubrežnih bolesnika je karakteristično da se
kalcifikacije razvijaju na 2 različita mesta u zidu krvnog suda: kao
kalcifikacije intimalnih plakova i kao medijalne kalcifikacije
ograničene na sloj glatkih mišićnih ćelija sve do lamine elastike
arterijskog zida. Kalcifikacije u intimi se obično javljaju kod
starijih dijaliznih bolesnika sa dužom istorijom netradicionalnih
faktora rizika (pušenje, dislipidemija), dok se medijalne
kalcifikacije javljaju kod bolesnika u proseku mlađih, oko 20
godina, sa dužim dijaliznim stažom i češćim poremećajima balansa Ca
i P. Vaskularne kalcifikacije su povezane sa KV mortalitetom preko
povećane brzine pulsnog talasa aorte koji povećanjem afterload-a
doprinosi povećanju mase leve komore [30]. Sa druge strane,
kalcifikacije valvula predstavljaju rizik za nastanak valvularne
stenoze, povećavaju rizik od pojave infektivnog endokarditisa,
hipetrofije leve komore i srčane slabosti, i takođe su nezavisni
prediktor kardiovaskularnog mortaliteta [31]. Mehanizam nastanka
kardiovaskularnih kalcifikacija u uremiji je složen i rezultat je
poremećenog mineralnog i koštanog metabolizma. Kalcijum fosfat se
taloži u krvnim sudovima, miokardu i srčanim valvulama uglavnom u
obliku apatita, ali to nije pasivan proces koji se zasniva na
precipitaciji viška Ca i P u estracelularnom matriksu, već se radi o
strogo kontrolisanom, aktivnom ćelijskom procesu koji uključuje
apoptozu, osteohondrogenu diferencijaciju glatkih mišićnih ćelija i
degradaciju elastina [32]. Čitav proces je regulisan interakcijom
brojnih modulatora: inhibitora kalcifikacija (fetuin A, matrix Gla
protein, ostepontin, osteoprotegerin, pirofosfati), promotera
kalcifikacija (fosfati, kalcijum, lipidi, inflamacija) i produkta
kalcijum x fosfor, paratireoidnog hormona i leptina [30, 32]. Sa
pogoršanjem bubrežne funkcije u HBB smanjuje se raspoloživost
aktivnog oblika vitamina D, i to usled nedostatka njegovog
prekursora, snižene aktivnosti 1 alfa hidroksilaze koja se sintetiše
u bubrezima i odgovorna je za konverziju prekursora u aktivni oblik
vitamina, ili oba faktora. Deficit vitamina D je povezan sa
povećanim rizikom KV događaja i promenom srčane strukture i funkcije
[13]. Vitamin D i PTH regulišu nivo neorganskog fosfata koji se,
zbog povećane senzitivnosti glatkih mišićnih ćelija, danas smatra
glavnim promoterom kalcifikacija u HBB. Neogranski fosfat nastaje
hidrolizom ekstracelularnog pirofosfata pomoću enzima alkalne
fosfataze čiji je nivo povišen u HBB. Ekstracelularni pirofosfat je
potentni inhibitor depozita kalcijum-fosfora in vitro i in vivo, i u
budućnosti bi mogao predstavljati novu terapijsku opciju za bubrežne
bolesnike. Aktuelna istraživanja se, generalno, više bave
inhibitorima kalcifikacija, jer je sinteza inhibitora osnovni
mehanizam kojim same ćelije mogu prevenirati razvoj vaskularnih
kalcifikacija [33].
Dislipidemija je jedan od najsnažnijih faktora rizika za neželjene
KV događaje u populaciji bolesnika sa HBB. Lipidni profil bolesnika
sa HBB se karakteriše niskim nivoom HDL holesterola i visokim nivoom
triglicerida, sa normalnim do niskim ukupnim holesterolom i LDL
holesterolom. Hipertrigliceridemija se pripisuje odloženom
katabolizmu i povećanoj hepatičkoj produkciji lipoproteina bogatih
trigliceridima, a u manjem stepenu prisustvu inhibitora lipaze. Mada
povećan nivo LDL holesterola nije tipična karakteristika ovih
pacijenata, jedna subfrakcija LDL-a niske gustine (sdLDL –
small-density) je povećana i ima sposobnost penetracije u zid krvnog
suda gde podleže ekscesivnoj oksidaciji i podstiče aterosklerotični
proces [34]. Osim toga, sastav i funkcija HDL holesterola su
značajno izmenjeni u HBB, što vodi ne samo gubitku zaštitnih
svojstava, već i transformaciji u toksičke partikule [35].
Zahvaljujući novostečenom aterogenom potencijalu, povećani nivo HDL
holesterola paradoksalno postaje tokom vremena značajno povezan sa
ukupnim i KV mortalitetom dijaliznih bolesnika [36].
Inflamacija niskog stepena je jedan od netradicionalnih faktora
rizika koji su ključni u patogenezi ateroskleroze, vaskularnih
kalcifikacija i drugih uzroka KVB, a može doprineti razvoju
proteinsko energetske malnutricije i drugih komplikacija u HBB.
Inflamatorni biomarkeri, kao što su visoko senzitivni CRP i
interleukin 6, nezavisno predviđaju mortalitet kod ovih bolesnika.
Uzroci inflamacije u HBB su multifaktorijalni i uključuju disbalans
između povećane produkcije (usled multiplih izvora inflamatornih
stimulusa kao što su oksidativni stres, acidoza, hipervolemija,
komorbiditeti, infekcije, genetski i epigenetski uticaji, i
dijalizna procedura) i neadekvatnog uklanjanja (usled snižene
glomerularne filtracije, ili neadekvatnog dijaliznog klirensa u
terminalnoj HBB) proinflamatornih citokina [37].
Ovo nisu jedini patofiziološki mehanizmi koji povezuju HBB sa
povećanim KV rizikom, ali mnogi od njih se tek otkrivaju ili se i
dalje istražuju, pa lista još uvek nije kompletna. Kompleksna
interakcija između srca i bubrega je najbolje opisana terminom
kardiorenalnog sindroma, koji obuhvata 5 tipova. Tip 4 se definiše
kao HBB koja dovodi do srčane hipertrofije, snižene renalne funkcije
i povećanog rizika neželjenih KV događaja, i u poslednje vreme se
često naziva „renokardijalni sindrom“ [38]. Rizik od KV događaja u
HBB je uplivisan, pored tradicionalnih faktora rizika, i specifičnim
faktorima rizika. Kako HBB progredira, tako faktori rizika
specifični za uremiju postaju sve dominantniji, a rizik od KVB
nezaustavljivo raste.
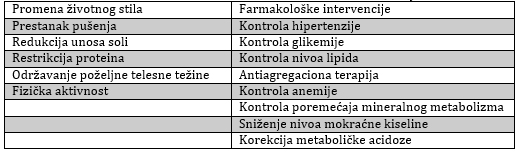
PREVENCIJA KARDIOVASKULARNIH BOLESTI
Prevencija KVB može se postići promenama životnog stila i
farmakološkim intervencijama. Kod bolesnika sa HBB, mere prevencije
su uglavnom usmerene na postizanje najbolje moguće kontrole
tradicionalnih faktora rizika. Međutim, s obzirom da se KV rizik
progresivno povećava sa opadanjem bubrežne funkcije, prevenciju
gubitka bubrežne funkcije treba postaviti kao cilj per se.
Strategije usmerene na usporavanje ili zaustavljanje progresivnog
gubitka bubrežne funkcije ne odlažu samo potrebu za terapijom zamene
bubrežne funkcije, već takođe smanjuju i KV rizik (tabela 1.).
Tabela 1. Prevencija kardiovaskularnih bolesti u
hroničnoj bubrežnoj bolesti
Table 1. Prevention of cardiovascular disease in chronic kidney
disease
PROMENE ŽIVOTNOG STILA
U opštoj populaciji pušenje je povezano sa rizikom razvoja HBB, dok
je u populaciji bubrežnih bolesnika udruženo sa ubrzanim opadanjem
JGF. Smatra se da pušenje podstiče razvoj fibroze bubrega
zahvaljujući negativnom uticaju na brojne faktore uključene u
fibrogenezu kao što su endotelna funkcija, oksidativni stres,
aktivacija faktora rasta uključujući angiotenzin II i endotelin-1,
pogoršanje metabolizma lipoproteina i insulinska rezistencija [39].
Rizik razvoja HBB opada sa vremenom koje prolazi od prestanka
pušenja i povećava sa kumulativnim izlaganjem, što sugeriše doznu
zavisnost i uzročno-posledičnu povezanost [40]. Pored negativnog
uticaja na progresiju HBB, pušenje doprinosi i povećanom KV riziku u
ovoj populaciji. U japanskoj kohortnoj studiji u kojoj je
učestvovalo skoro 35000 osoba, kod bolesnika sa HBB pušenje je bilo
udruženo sa duplo većim rizikom ukupnog i KV mortaliteta [41]. Sve
je više dokaza o korisnim efektima prekida pušenja, pa je nedavno
saopšteno da prekid pušenja smanjuje opadanje JGF kod nedijabetičnih
bolesnika sa HBB lečenih inhibitorima angiotenzin konvertujućeg
enzima (ACEi) [42]. Stoga, prekid pušenja treba stimulisati i
ohrabrivati kako kod osoba sa HBB, tako i kod onih sa prisutnim
faktorima rizika za razvoj HBB.
Bolesnici sa HBB se odlikuju visokom učestalošću hipertenzije koja
je senzitivna na so i sklonošću ka retenciji tečnosti. Visoki unos
soli može podstaći renalnu disfunkciju i progresiju HBB putem
direktnih i indirektnih mehanizama: povećanja ekstracelularnog
volumena, glomerularne hiperperfuzije koja rezultira
glomerulosklerozom i proteinurijom, stimulacije sinteze
proinflamatornih citokina, povećanja oksidativnog stresa, aktivacije
simpatičke aktivnosti. Nedavno je dokazano je da visoki unos soli
može oslabiti antihipertenzivne i antiproteinurične efekte blokatora
RAAS [43]. Povezanost unosa soli sa krvnim pritiskom je osnova
verovanja da restrikcija unosa soli može prevenirati KV događaje
povezane sa hipertenzijom, pa se stoga preporučuje da dnevni unos
kuhinjske soli u HBB bude 2000mg/dan a da ne premašuje 5g dnevno
[44]. Međutim, povezanost između unosa soli i KV rizika nije
definitivno razjašnjena. Tako je nedavna analiza 4 velike studije
pokazala da je kod hipertenzivnih bolesnika ekskrecija Na veća od
7g/dan, kao marker unosa soli, povezana sa povećanim rizikom KVB,
ali je i ekskrecija Na manja od 3g/dan takođe bila udružena sa
povećanim KV rizikom [45].
Poznato je da dijeta sa visokim sadržajem proteina nepovoljno utiče
na renalnu hemodinamiku povećanjem intraglomerulskog pritiska i
glomerulskom hiperfiltracijom, što dugoročno može dovesti do
oštećenja bubrežne funkcije, naročito u uslovima već postojećeg
oboljenja bubrega. Međutim, uprkos brojnim kliničkim studijama
efikasnost niskoproteinske dijete u usporavanju progresije HBB
ostaje i dalje kontroverzna. Trenutno je aktuelno gledište po kome
niskoproteinska dijeta pokazuje korisne efekte na smanjenje rizika
od razvoja terminalnog stadijuma HBB usled smanjenja uremijskih
simptoma i odlaganja dijalizne terapije, uprkos malom efektu na
opadanje JGF. Niskoproteinska dijeta takođe smanjuje metaboličku
acidozu i hiperfofatemiju, čime se postiže bolja kontrola poremećaja
koštanog i mineralnog metabolizma i ublažava insulinsku rezistenciju
i oksidativni stres [46]. Uzimajući u obzir ove korisne efekte, sa
jedne strane, i rizik od malnutricije izražen kod starijih osoba sa
druge strane, aktuelne smernice sugerišu proteinski unos niži od
0,8g/kg telesne težine za odrasle bubrežne bolesnike čija je JGF
manja od 30mL/min/1,73m2. Implementaciju ovih preporuka je teško
sprovesti u praksi zbog nedostatka komplijanse i potrebe za strogim
nadzorom kod velikog broja bolesnika sa HBB [44]. Stoga, najnovije
preporuke radne grupe ERA/ EDTA (European Renal Association/
European Dialysis and Transplantation Association) o renalnoj
nutriciji predlažu 5 (stadijum 3b HBB) do 8 poseta (stadijum 4 HBB)
dijetetičaru godišnje, sa ciljem da se postigne optimalni nadzor
koji bi omogućio individualni pristup navikama svakog bolesnika
[47].
Gojaznost je nezavistan faktor rizika za razvoj i progresiju HBB.
Ona može indirektno voditi do HBB povećanjem rizika od hipertenzije,
ateroskleroze i diabetes mellitus-a tip 2, ali može imati i direktne
patofiziološke efekte: kroz glomerulsku hiperfiltraciju, faktore
rasta i promene nivoa adipokina može voditi fibrozi,
glomerulosklerozi i bubrežnoj bolesti [48]. Pored toga, utvrđeno je
da gojaznost ima prediktivnu vrednost za KV bolest i mortalitet kod
bolesnika sa HBB, pri čemu se obim struka pokazao kao bolji
parametar za procenu rizika od BMI [49]. Sistematski pregled 13
randomizovanih, kontrolisanih studija o gubitku težine kod gojaznih
osoba sa HBB, otkriva da je namerni gubitak težine bio povezan sa
redukcijom proteinurije nezavisno od efekta na sniženje krvnog
pritiska [50]. Sa druge strane, neke studije su pokazale da je
gojaznost protektivni faktor kod dijaliznih bolesnika sa terminalnim
stadijumom HBB, verovatno zato što je malnutricija udružena sa višim
mortalitetom u poređenju sa gojaznošću. Međutim, čak i kod dijalizno
lečenih bolesnika, visceralna gojaznost je udružena sa povećanim
rizikom koronarnih kalcifikacija i neželjenih kardiovaskularnih
događaja [51].
Između gojaznosti i smanjene fizičke aktivnosti postoji snažna veza.
Svetska zdravstvena organizacija je nedavno saopštila da je fizička
neaktivnost četvrti vodeći uzrok mortaliteta u svetu, što ukazuje da
sedanterni način života treba posmatrati kao zasebnu bolest [52].
Bolesnici sa HBB se odlikuju smanjenom tolerancijom napora i
smanjenom fizičkom aktivnošću. Opservacione studije su pokazale da
je povećanje fizičke aktivnosti kod bolesnika sa HBB udruženo sa
sniženjem albuminurije [53] i ukupnog i KV mortaliteta [54]. Osim
toga, fizička aktivnost umerenog intenziteta je povezana sa
poboljšanjem fizičkih performansi, poboljšanjem kvaliteta života i
redukcijom KV faktora rizika. Stoga smernice preporučuju da se osobe
sa HBB podstiču na fizičku aktivnost koja je kompatibilna sa
kardiovaskularnim zdravljem i tolerancijom (sa ciljem najmanje po 30
minuta 5 puta nedeljno) i na postizanje zdrave telesne težine (BMI
20 do 25, prema demografiji specifičnoj za pojedinu zemlju) [44].
FARMAKOLOŠKE INTERVENCIJE
Antihipertenzivna terapija
Hipertenzija kod bubrežnih bolesnika poseduje neke specifičnosti o
kojima treba voditi računa u cilju postizanja optimalne kontrole KP.
Kod zdravih osoba, KP pokazuje noćni pad od približno 10-20%, koji
je regulisan delovanjem nekoliko faktora uključujući diurnalne
varijacije u autonomnoj funkciji nervnog sistema, ekskreciju soli i
RAAS. Poremećena regulacija ovih sistema u HBB vodi „non-dipping-u“
ili čak porastu KP u toku noći, koji je udružen sa povećanim KV
morbiditetom i mortalitetom i rizikom progresije HBB [55]. Adekvatno
sniženje KP redukuje proteinuriju čime usporava opadanje JGF i
smanjuje rizik KV događaja. Intenzivnija redukcija krvnog pritiska
(ciljni sistolni KP <120 mmHg) može pružiti veću renoprotekciju
osobama sa značajnom proteinurijom [>1 g/dan; PCR
(protein-to-creatinin ratio) >100mg/mmol, ACR (albumin-to-creatinine
ratio) >70mg/mmol] u odnosu na one bez proteinurije. To ukazuje da
je pored antihipertenzivnog efekta, u izboru optimalne terapije kod
bolesnika sa HBB važno uzeti u obzir i uticaj lekova na proteinuriju
[56]. Smernice objavljene od strane American College of Cardiology
(ACC) 2017. godine preporučuju ciljne vrednosti KP niže od
130/80mmHg za sve odrasle osobe sa hipertenzijom i HBB, bez obzira
na protenuriju [57]. The National Institute for Health and Care
Excellence (NICE) i UK Renal Association sugerišu konzervativniji
pristup ciljnim vrednostima KP, koje bi trebalo da budu niže od
140/90 mmHg ukoliko je proteinurija manja od 1,0 g/dan i niže od
130/80 mmHg u prisustvu većeg stepena proteinurije [58, 59]. KDIGO
(Kidney Disease Improving Globale Outcomes) smernice takođe sugerišu
niže vrednosti ciljnog KP za one sa značajnom proteinurijom,
definisano vrednošću >300 mg/dan [60]. ESC/ESH (European Society of
Cardiology/European Society of Hypertension) smernice sugerišu
ciljni sistolni KP <140mmHg, bez obzira na stepen proteinurije [61].
Očigledno je da postojeći dokazi iz različitih studija ne obezbeđuju
jasan konsenzus u pogledu optimalnih vrednosti ciljnog krvnog
pritiska u HBB, a u skladu sa dokazima variraju i preporuke koje se
navode u smernicama. Na osnovu do sada izloženih stavova moglo bi se
zaključiti da bi za prevenciju KVB verovatno bilo dovoljno
održavanje vrednosti KP ispod 140/90mmHg. Međutim, optimalna
renoprotekcija se postiže sa vrednostima KP nižim od 130/80mmHg,
naročito kod bubrežnih bolesnika sa većim stepenom albuminurije ili
dijabetesnom nefropatijom [13]. Lečenje visokog krvnog pritiska kod
bolesnika sa bilo kojim stadijumom HBB je od ogromne važnosti za
usporavanje ili prevenciju progresije HBB, i predstavlja osnovu
kardiovaskularne protekcije. Iako bilo koja klasa antihipertenzivnih
lekova može biti efikasna kod bubrežnih bolesnika, od kada je
utvrđeno da blokada RAAS redukuje proteinuriju nezavisno od KP, ovi
lekovi se smatraju prvom terapijskom linijom za bolesnike sa
mikroalbuminurijom i proteinurijom [56, 61]. Primena RAAS blokatora
je pokazala korisne efekte na redukciju ukupnog i KV mortaliteta,
odnosno na bolje preživljavanje bolesnika u različitim stadijumima
HBB [14]. Izbor dodatnih lekova za sniženje krvnog pritiska treba
uskladiti sa potrebama i tolerancijom individualnih pacijenata.
Najnovije smernice ESC/ESH kao inicijalnu terapiju sugerišu
kombinaciju RAAS blokatora sa kalcijumskim antagonistima ili
diureticima, dok se kombinacija 2 RAAS blokatora ne preporučuje
[61]. Bolesnici sa HBB su obično u hipervolemiji i diuretska
terapija je često indikovana. Sa opadanjem GFR, u 4. stadijumu HBB,
diuretici Henleove petlje zamenjuju tiazide i postaju neophodni za
kontrolu retencije tečnosti i pridružene hipertenzije [44].
Diuretici povećavaju efikasnost RAAS inhibitora u sniženju
albuminurije [13], što može rezultirati u dodatnoj renoprotekciji.
Tretman beta blokatorima redukuje ukupni mortalitet kod bolesnika sa
HBB i hroničnom sistolnom srčanom insuficijencijom kroz sve
stadijume predijalizne HBB [62]. Rezultati dosadašnjih istraživanja
pokazali su da većina bolesnika ne uspeva da dostigne ciljne
vrednosti krvnog pritiska uprkos intenzivnoj kontroli,
nefarmakološkim intervencijama i višestrukoj antihipertenzivnoj
terapiji [56]. Postizanje ciljnih vrednosti krvnog pritiska je stoga
i dalje terapijski izazov, čak veći u populaciji bolesnika sa HBB
nego u opštoj populaciji.
Kontrola glikemije
Diabetes mellitus je vodeći uzrok HBB i nezavisan je faktor rizika
za KV mortalitet kod ovih bolesnika [44]. Tokom poslednjih 20 godina
razjašnjeno je da glukoza podstiče stvaranje reaktivnih kiseoničnih
radikala i da zajedno sa krajnjim produktima uznapredovale
glikozilacije dovodi do aktivacije intracelularnih signalnih
molekula, kao što su protein kinaza C i nuklearni faktor kapa B,
koji povećavaju ekspresiju faktora rasta i citokina, što rezultira
povećanom akumulacijom ekstracelularnog matriksa, pojavom
albuminurije i inflamacije u dijabetesnom bubregu. Osim metaboličkim
faktorima, ovi putevi se takođe aktiviraju i hemodinamskim faktorima
(sistemska hipertenzija, angiotenzin II), indukujući progresivno
oštećenje bubrega [63]. Logično bi bilo očekivati da adekvatna
kontrola hiperglikemije oslabi štetan učinak aktivacije tih
prosklerotičnih i proinflamatornih puteva u bubregu, i da smanji
funkcionalne i strukturne manifestacije dijabetesne nefropatije.
Postoje dokazi iz kliničkih studija da optimalna kontrola glikemije
usporava progresiju mikrovaskularnih komplikacija dijabetesa,
uključujući nefropatiju procenjenu albuminurijom [64], ali rezultati
meta analize pokazali su samo ograničenu korist od intenzivnog
tretmana hiperglikemije u poboljšanju KV ishoda ili smanjenju
mortaliteta [65].
Hipoglikemija se u brojnim studijama prepoznaje kao glavni rizik
previše intenzivne glikemijske kontrole, i taj rizik se povećava sa
sniženjem bubrežne funkcije. Stoga se u poslednjim KDIGO smernicama
preporučuje ciljna vrednost HbA1c od približno 7% za prevenciju ili
odlaganje progresije mikrovaskularnih komplikacija dijabetesa,
uključujući dijabetesnu bolest bubrega. Ukoliko postoji rizik od
hipoglikemije ciljni HbA1c ne sme biti <7%, a savetuje se da bude
iznad 7% i u slučaju prisutnih komorbiditeta ili ograničenog
očekivanog životnog veka. Kod bolesnika koji imaju HBB i DM,
kontrola glikemije čini deo preporučenih multifaktorijalnih
interventnih strategija usmerenih na kontrolu krvnog pritiska i
kardiovaskularnog rizika, što podrazumeva upotrebu ACEi, ARB,
statina i inhibitora agregacije trombocita gde je to klinički
indikovano [44].
Nedavno objavljena meta analiza o efektima oralnih hipoglikemika na
ukupni i kardiovaskularni mortalitet u dijabetesu tipa 2, pokazala
je da preparati sulfonil ureje značajno povećavaju rizik ukupnog
mortaliteta, dok inhibitori dipeptil peptidaze 4 (DPP4i) u
kombinaciji sa metforminom kao dvojna terapija konzistentno smanjuju
rizik ukupnog i KV mortaliteta [66]. Međutim, u uznapredovalom
stadijumu HBB metformin i dugo-delujući hipoglikemijski agensi su
kontraindikovani zbog rizika od hipoglikemije. Sa druge strane,
pojava novih klasa hipoglikemika, kao što su inhibitori Na glukoznog
kotransportera 2 (SGLT2) i agonisti glukagonu-sličnog peptid-1
(GLP-1) receptora rezultirala je impresivnim efektima na
kardiovaskularnu i renalnu funkciju. Iako mehanizam
kardioprotektivnog i renoprotektivnog delovanja ovih lekova nije u
potpunosti razjašnjen, ovi novi tretmani ulivaju optimizam kad je
reč o redukciji mikrovaskularnih i makrovaskularnih komplikacija kao
glavnih uzroka morbiditeta i prevremenog mortaliteta u dijabetesu
[67].
Kontrola dislipidemije
Terapija za sniženje lipida nesumnjivo doprinosi smanjenoj incidenci
KV događaja u opštoj populaciji, a slična korist je zabeležena i u
populaciji bolesnika sa HBB, posebno onih sa visokim nivoima
serumskog holesterola. Tako je u SHARP studiji (Study of Heart and
Renal Protection) koja je obuhvatila bolesnike u svim fazama HBB
uključujući 32% dijaliznih bolesnika, primena ezetimiba u
kombinaciji sa statinima dovela do prosečnog sniženja LDL
holesterola od 0,85 mmol/L, što je bila praćeno značajnom redukcijom
aterosklerotičnih događaja za 17% [68]. Međutim, meta analiza svih
studija sa statinima kod osoba sa HBB pokazuje da je KV protektivni
efekat ovih lekova oslabljen kod onih sa niskim vrednostima JGF i
ograničen kod bolesnika na hemodijalizi [69]. Malo je dostupnih
podataka koji ukazuju da snižavanje lipida utiče na brzinu
progresije bubrežne bolesti [13].
KDIGO smernice iz 2013. godine preporučuju lečenje statinima ili
kombinacijom statin/ezetimib kod svih odraslih osoba starijih od 50
godina koji imaju JGF nižu od 60 ml/min/1,73 m2, a još uvek se ne
leče ponavljanim dijalizama (stadijum 3a-5), kao i kod predijaliznih
bolesnika starosti između 18 i 49 godina ukoliko postoje drugi
faktori rizika (koronarna bolest, diabetes mellitus,
cerebrovaskularni insult). Štaviše, ove smernice ne preporučuju
započinjanje terapije statinima kod bolesnika na dijalizi, ali
predlažu nastavak terapije ukoliko bolesnik već uzima statine. Pri
tom tretman treba sprovoditi prema strategiji “ fire and forget”,
što znači da ne postoje određene ciljne vrednosti koje bi zahtevale
praćenje lipidnog profila. Ukoliko postoji hipertrigliceridemija,
savetuje se samo promena životnog stila. Prethodne smernice su
sugerisale upotrebu fibrata za prevenciju pankreatitisa u teškoj
hipertrigliceridemiji, ali dokazi koji podržavaju sigurnost i
efikasnost ovog pristupa su slabi, naročito kod bolesnika sa HBB.
Terapija fibratima (fenofibrat i gemfibrozil) može se uzeti u
razmatranje samo kod bolesnika sa trigliceridima >11.3 mmol/l, uz
prilagođavanje doze bubrežnoj funkciji. Kombinacija statina sa
fibratima se ne preporučuje zbog potencijalne toksičnosti [44].
Antiagregaciona i antikoagulantna terapija
Bolesnici sa HBB su izloženi većem riziku razvoja endotelne
disfunkcije i ateroskleroze [19, 20]. Povećani KV rizik ukazuje da
oni mogu imati korist od antiagregacione terapije, ali abnormalna
funkcija trombocita koja karakteriše HBB može povećati rizik od
hemoragijskih događaja tokom primene antikoagulantne ili
antitrombocitne terapije. Do danas su dostupni ograničeni podaci o
efikasnosti i bezbednosti antiagregacione terapije u HBB, pa
aktuelna saznanja primarno potiču iz meta analiza i pregleda
literature. Često citirani Cohranov sistematski pregled koji je
uključio bolesnike iz 50 studija u svim stadijumima HBB pokazao je
da antiagregaciona terapija redukuje rizik infarkta miokarda, ali ne
i rizik ukupnog mortaliteta, KV mortaliteta i moždanog udara, sa
povećanom incidencom velikih i malih epizoda krvarenja [70]. Rizik
od hemoragijskih manifestacija se povećava sa kombinovanom
antiagregacionom terapijom koja se primenjuje u akutnom koronarnom
sindromu. Od antiagregacionih lekova, klopidogrel je pokazao slabiju
efikasnost kod bolesnika sa HBB, tikagrelor je pokazao benefit u
odnosu na mortalitet bolesnika sa renalnom disfunkcijom, dok su
inhibitori glikoproteina IIb/IIIa (GPIs) pokazali benefit tretmana
sa rizikom od krvarenja. Modifikacija doze GPIs je neophodna kod
bolesnika sa bubrežnom slabošću čime se redukuje rizik od krvarenja
[14]. Iznenađujuća opservacija je da kod bolesnika sa HBB i
atrijalnom fibrilacijom antikoagulans varfarin može povećati rizik
od moždanog udara, što se objašnjava potenciranjem vaskularnih i
valvularnih kalcifikacija (71).
Aktuelne smernice preporučuju niske doze aspirina (75-100mg dnevno)
kod muškaraca starosti između 45 i 79 godina i žena starosti između
55 i 79 godina, ako korist od prevencije KVB nadmašuje rizik
neželjenih događaja, uključujući gastrointestinalna krvarenja [72].
Druge smernice preporučuju niske doze aspirina (75-162mg dnevno) za
bolesnike sa istorijom KVB ili niske doze aspirina u kombinaciji sa
tienopiridinima za bolesnike sa istorijom perkutane kornonarne
intervencije ili stenta [73]. Za bolesnike sa jednim ili više KV
faktora rizika (diabetes, hiperlipidemija), koristi od uzimanja
aspirina mogu nadmašiti rizike. Međjutim, korist od antiagregacione
i antikoagulantne terapije za primarnu i sekundarnu prevenciju kod
bolesnika sa HBB ostaje neizvesna, dok je rizik od krvarenja
povećan, pa donošenje odluke o terapiji zavisi od induvidualne
procene lekara. S obzirom na nesigurnu korist i mogućnost štete,
trenutne smjernice KDIGO preporučuju da se aspirin ne koristi za
primarnu prevenciju KVB kod bolesnika s HBB [44].
Lečenje anemije
Anemija u HBB je povezana sa rizikom KV komplikacija, bržom
progresijom HBB i smanjenim kvalitetom života. Od KV komplikacija,
anemija je povezana sa razvojem HLK koja se nalazi kod 74% bolesnika
na početku dijaliznog lečenja i nezavisan je prediktor kardijalnog
morbiditeta i mortaliteta. HLK može doprineti razvoju hronične
srčane insuficijencije, koja dovodi do opadanja bubrežne perfuzije,
rezultirajući u daljem bubrežnom oštećenju (tzv. cardiorenal anemia
syndrome). Osim toga, anemija je jedan od najčešćih razloga
ponavljanih hospitalizacija bubrežnih bolesnika sa rekurentnim
epizodama kongestivne srčane insuficijencije i pogoršanjem angine
pectoris [2, 74, 75]. Iz ovoga je proisteklo logično očekivanje da
će adekvatno lečenje anemije poboljšati KV status bolesnika sa HBB.
Ciljni nivo Hb koji treba dostići lečenjem dugo je bio predmet
debate i inicijalno se težilo normalizaciji Hb. Početne male
opservacione studije su pokazale bolji ishod sa višim ciljnim
vrednostima Hb, ali kasnije sprovedene interventne studije sa
kompletnom korekcijom anemije nisu dale očekivane rezultate.
Najpoznatije su dve velike randomizovane kontrolisane studije CREATE
(Cardiovascular reduction Early Anemia Treatment Epoetin β) i CHOIR
(Correction of Hemoglobin and Outcomes in Renal Insufficiency), od
kojih prva nije dovela do redukcije neželjenih ishoda, dok je druga
pokazala značajan porast u KV događajima i mortalitetu sa porastom
ciljnih vrednosti Hb [76, 77]. TREAT (Trial to Reduce Cardiovascular
Events With Aranesp Therapy) studija je kasnije pokazala da primena
Darbepoetina alfa nije redukovala rizik od definisanih kompozitnih
događaja, a bila je povezana sa povećanim rizikom cerebrovaskularnog
insulta [78]. Rukovodeći se ovim rezultatima, KDIGO smernice
preporučuju da kod odraslih bolesnika sa HBB nivo Hb ne bi trebalo
da premaši 11.5 g/dL, mada ta granica može biti liberalna ukoliko se
bolesnik oseća bolje sa višim vrednostima, a spreman je da prihvati
rizike. Rizik od hipertenzije, kongestivne srčane insuficijencije i
moždanog udara se povećava kada Hb premaši granicu od 12g/dL. Stoga,
za optimalno KV i opšte zdravlje, terapiju treba titrirati kako bi
se postigao ciljni Hb u rasponu od 10-12 g/dL [14, 44].
Korekcija poremećaja mineralnog metabolizma
Sekundarni hiperparatireoidizam je centralna komponenta u
poremećajima mineralno koštanog metabolizma u HBB i povezan je sa
povećanim KV morbiditetom i mortalitetom [76]. Vrlo rano tokom
progresije bubrežne slabosti, pre porasta PTH raste nivo FGF23 i
povećane koncentracije FGF23 su nezavisno povezane sa povećanim
rizikom progresije HBB, KV komplikacija i mortaliteta u HBB [79,
80]. FGF23 je hormon koji reguliše nivo fosfata u organizmu, a
hiperfosfatemija je, takođe, udružena sa povećanim rizikom ukupnog
mortaliteta i KV događjaja, iako se pretpostavlja da FGF23 i
hiperfosfatemija utiču na ishod različitim mehanizmima KV
toksičnosti [81].
Prema aktuelnim KDIGO smernicama, preporučuje se skrining
sekundarnog hiperparatiroidizma kod bolesnika sa HBB kada JGF opadne
ispod 45ml/min/1,73m2, merenjem serumskog nivoa Ca, P, PTH i alkalne
fosfataze da bi se determinisale bazalne vrednosti. Savetuje se
održavanje serumskih vrednosti P u normalnom rangu prema referentnim
vrednostima lokalne laboratorije. Ako je nivo PTH iznad gornje
granice, te osobe se prvo procenjuju za hiperfosfatemiju,
hipokalcemiju i deficit vitamina D. Za supresiju povišenih
koncentracija PTH kod predijaliznih bolesnika, ne sugeriše se
rutinsko propisivanje vitamina D ili vitamin D analoga u odsustvu
suspektnog ili dokumentovanog deficita. Takođe, ne savetuje se
propisivanje bifosfonata kod osoba sa GFR <30 ml/min/1,73m2, bez
jakih kliničkih razloga [44].
U kontroli hiperfosfatemije kod bolesnika sa HBB koristi se dijeta
sa niskim sadržajem fosfora (preporučeni dnevni unos je
800-1000mg/dan) i vezivači fosfata. Danas je dostupan veliki broj
vezivača fosfata na svetskom tržištu, uključujući aluminijum
hidroksid, kalcijumove soli, sevelamer hidrohlorid, sevelamer
karbonat, lantanum karbonat i vezivače na bazi gvožđa [79]. U
prospektivnoj kohortnoj studiji koja je obuhvatila 10044 bolesnika
na hemodijalizi, tretman vezivačima fosfata je bio nezavisno povezan
sa smanjenim mortalitetom [82]. Vitamin D se propisuje kod bolesnika
sa HBB u cilju supresije PTH i to uglavnom u aktivnoj formi kao
kalcitriol, parikalcitol ili doxercalciferol [79]. Randomizovane
studije su pokazale korisne efekte parikalcitola na albuminuriju,
međutim u kasnijim studijama parikalcitol nije pokazao pozitivan
uticaj na srčanu strukturu i funkciju kod bolesnika sa HBB. Stoga su
neophodne veće studije sa dužim periodom praćenja, a suplementacija
vitamina D još uvek nije indikovana za smanjenje KV rizika u HBB
[79, 13]. Sinakalcet hidrohlorid je kalcimimetički agens koji
uspešno suprimira sekreciju PTH, a krajnji rezultati randomizovane
kontrolisane studije koja je uključila bolesnike na HD sa umerenim
do teškim hiperparatireoidizmom pokazali su da sinakalcet značajno
smanjuje rizik KV događaja [83]. U međuvremenu je zabeleženo da
bolesnici koji uzimaju sinakalcet imaju povećanu učestalost mučnine,
povraćanja i hipokalcemije, tako da potencijalni rizici primene
Sinakalceta mogu premašiti korist [79]. Uprkos maksimalnoj
medikamentnoj terapiji, hirurška paratireoidektomija je još uvek
neophodna za mnoge bolesnike.
Kontrola hiperurikemije
U opštoj populaciji, hiperurikemija je povezana sa povećanim rizikom
brojnih komplikacija, uključujući razvoj KVB i HBB. Kod bolesnika sa
uznapredovalom HBB, podaci iz opservacionih studija nisu dokazali
povezanost nivoa mokraćne kiseline sa progresijom HBB, iako je
potvrđena korelacija sa endotelnom disfunkcijom, ukupnim i KV
mortalitetom. Sa druge strane, male interventne studije su pokazale
da lečenje asimptomatske hiperurikemije kod bolesnika sa HBB
usporava progresiju HBB, poboljšava endotelnu funkciju i elastičnost
centralnih arterija, redukuje masu leve komore i KV događaje [44].
Očigledno je da postoji mnogo kontradiktornih podataka o ulozi
mokraćne kiseline i njenoj povezanosti sa KV događajima i ukupnim
mortalitetom kod bolesnika sa uznapredovalom HBB. Poslednjih godina
se povezanost izmeđju mokraćne kiseline i ukupnog i KV mortaliteta
sve češće opisuje krivom J oblika, što se objašnjava ulogom mokraćne
kiseline kao nutritivnog markera koji prevashodno ispoljava
antioksidantna svojstva u ovoj populaciji bolesnika [84]. Stoga,
aktuelne KDIGO smernice priznaju da je nedovoljno dokaza koji bi
podržali ili osporili upotrebu agenasa za sniženje koncentracije
mokraćne kiseline u serumu kod bolesnika sa HBB i simptomatskom ili
asimptomatskom hiperurikemijom u cilju usporavanja progresije HBB i
ističu potrebu za novim velikim studijama koje bi ispitale
potencijalnu korist ovih lekova [44].
Korekcija acidoze
Metabolička acidoza kao komplikacija uznapredovalog stadijuma HBB
udružena je sa malnutricijom, inflamacijom, metaboličkom bolešću
kostiju, ubrzanom progresijom HBB i povećanim mortalitetom [80, 44].
Velike acido-bazne oscilacije tokom dijalize i njima intenzivirani
poremećaji nivoa Ca mogu igrati ulogu u razvoju KVB kod
hemodijaliznih bolesnika. Nedavno je potvrđena značajna povezanost
metaboličke acidoze sa prisustvom periferne vaskularne bolesti i
dijastolne disfunkcije kod bolesnika na intermitentnoj dijalizi
[85]. U drugoj prospektivnoj studiji dokazana je statistički
značajna povezanost između nivoa dijaliznih bikarbonata, parametara
mineralnog metabolizma (Ca i P), brzine pulsnog talasa kao parametra
vaskularne rigidnosti i broja kalcifikacija, što odražava KV rizik
[86]. Kod bolesnika sa HBB i serumskom koncentracijom HCO3 <22
mmol/l, aktuelne KDIGO smernice preporučuju tretman oralnim
suplementima kako bi se serumski bikarbonati održali unutar
normalnog ranga, ukoliko ne postoje kontraindikacije [44].
ZAKLJUČAK I PERSPEKTIVE
Od prvih opservacija o povezanosti HBB i KVB koje je zabeležio
Richard Biright, do današnjih dana, prešli smo dugi put. Mnoge
epidemiološke studije potvrdile su ovu povezanost i ustanovile da
ona nije samo posledica zajedničkih faktora rizika kao što su
pušenje, gojaznost, hipertenzija, hiperholesterolemija i dijabetes.
Vremenom je utvrđeno da mehanizmi specifični za HBB podstiču i
razvoj vaskularne bolesti i na taj način povećavaju učestalost KVB
kod osoba sa HBB.
Nedovoljno poznavanje patofizioloških mehanizama je jedan od razloga
zbog kojih je KVB kod bubrežnih bolesnika još uvek nedovoljno
dijagnostikovana i lečena, uprkos dostupnosti dokazano efikasnih
tretmana. S obzirom da se bolesnici sa HBB smatraju grupom najvišeg
rizika za razvoj KVB, pored tradicionalnih, dobro poznatih, faktora
rizika važno je identifikovati i netradicionalne faktore rizika koji
nisu uključeni u postojeće KV prognostičke modele, kako bi se
omogućio razvoj budućih KV prognostičkih modela koji bi bili
specifični za HBB. Posebnu pažnju bi trebalo usmeriti na
istraživanje preventivnih strategija u ranim stadijumima bubrežne
slabosti, kao i na evaluaciju multifaktorskih intervencija u
uznapredovalom stadijumu HBB.
LITERATURA
- Bright R. Cases and observations illustrative of renal
disease accompanied with the secretion of albuminous urine.
Guy’s Hospital Trans. 1836; 1:338–379.
- Tirmenštajn-Janković Biserka, Dimković N. Faktori rizika
kardiovaskularnog oboljevanja u hroničnoj bubrežnoj bolesti.
Zvezdara Clin Proc 2007; 8(1-2): 33-42.
- Lindner A, Charra B, Sherrard DJ, Scribner BH. Accelerated
atherosclerosis in prolonged maintenance hemodialysis. N Engl J
Med 1974; 290: 697-701.
- Foley RN, Parfrey PS, Sarnak MJ. Epidemiology of
cardiovascular disease in chronic renal disease. J Am Soc
Nephrol 1998; 9 (12 Suppl): S16-23.
- Matsushita K van der Velde M Astor BC, Woodward M, Levey AS,
de Jong PE, et al; Chronic Kidney Disease Prognosis Consortium.
Association of estimated glomerular filtration rate and
albuminuria with all-cause and cardiovascular mortality in
general population cohorts: a collaborative meta-analysis.
Lancet. 2010; 375: 2073-2081.
- Van der Velde M, Matsushita K, Coresh J, Astor BC, Woodward
M, Levey A, et al; Chronic Kidney Disease Prognosis Consortium.
Lower estimated glomerular filtration rate and higher
albuminuria are associated with all-cause and cardiovascular
mortality. A collaborative meta-analysis of high-risk population
cohorts. Kidney Int. 2011; 79: 1341-1352.
- Hemmelgarn BR Clement F Manns BJ, Klarenbach S, James MT,
Ravani P et al. Overview of the Alberta Kidney Disease Network.
BMC Nephrol. 2009; 10: 30.
- Gansevoort RT, Matsushita K, van der Velde M, Astor BC,
Woodward M, Levey AS, et al; Chronic Kidney Disease Prognosis
Consortium. Lower estimated GFR and higher albuminuria are
associated with adverse kidney outcomes. A collaborative
meta-analysis of general and high-risk population cohorts.
Kidney Int 2011; 80: 93-104.
- Mahmoodi BK, Matsushita K, Woodward M, Blankestijn PJ,
Cirillo M, Ohkubo T, et al. for the Chronic Kidney Disease
Prognosis Consortium. Associations of kidney disease measures
with mortality and end-stage renal disease in individuals with
and without hypertension: a meta-analysis. Lancet 2012;
380(9854): 1649-61.
- Fox CS, Matsushita K, Woodward M, Bilo HJ, Chalmers J,
Heerspink HJ, et al. for the Chronic Kidney Disease Prognosis
Consortium. Associations of kidney disease measures with
mortality and end-stage renal disease in individuals with and
without diabetes: a meta-analysis. Lancet
2012;380(9854):1662-73.
- Matsushita K, Coresh J, Sang Y, Chalmers J, Fox C, Guallar
E, et al. for the CKD Prognosis Consortium. Estimated glomerular
filtration rate and albuminuria for prediction of cardiovascular
outcomes: a collaborative meta-analysis of individual
participant data. Lancet Diabetes Endocrinol 2015; 3(7): 514-25.
- Schiffrin EL, Lipman ML, Mann JF. Chronic kidney disease:
effects on the cardiovascular system. Circulation
2007;116(1):85-97.
- Gansevoort RT, Correa-Rotter R, Hemmelgarn BR, Jafar TH,
Heerspink HJ, Mann JF, et al. Chronic kidney disease and
cardiovascular risk: epidemiology, mechanisms, and prevention.
Lancet 2013; 382(9889): 339-52.
- Subbiah AK, Chhabra YK, Mahajan S. Cardiovascular disease in
patients with chronic kidney disease: a neglected subgroup.
Heart Asia 2016; 8(2): 56-61.
- Converse RL Jr, Jacobsen TN, Toto RD, Jost CM, Cosentino F,
Fouad-Tarazi F, et al. Sympathetic overactivity in patients with
chronic renal failure. N Engl J Med 1992; 327(27): 1912-8.
- Van Buren PN, Inrig JK. Hypertension and hemodialysis:
pathophysiology and outcomes in adult and pediatric populations.
Pediatr Nephrol 2012; 27(3): 339–50.
- Zoccali C Mallamaci F Maas R for the CREED Investigators.
Left ventricular hypertrophy, cardiac remodeling and asymmetric
dimethylarginine (ADMA) in hemodialysis patients. Kidney Int
2002; 62: 339-345.
- Kielstein JT Zoccali C. Asymmetric dimethylarginine: a
cardiovascular risk factor and a uremic toxin coming of age? Am
J Kidney Dis 2005; 46: 186-202.
- Dhaun N, Goddard J, Webb DJ. The endothelin system and its
antagonism in chronic kidney disease. J Am Soc Nephrol 2006;
17(4): 943-55.
- Kim ED, Tanaka H, Ballew SH, Sang Y, Heiss G, Coresh J,
Matsushita K. Associations Between Kidney Disease Measures and
Regional Pulse Wave Velocity in a Large Community-Based Cohort:
The Atherosclerosis Risk in Communities (ARIC) Study. Am J
Kidney Dis 2018; 72(5): 682-90.
- Monhart V. Education in Cardiology. Hypertension and chronic
kidney diseases. Cor et Vasa 2013; 55 (4): e397-e402
- Levin A, Singer J, Thompson CR, Ross H, Lewis M. Prevalent
LVH in the predialysis population: Identifying opportunities for
intervention. Am J Kidney Dis 1996; 27: 347-54.
- Foley, R. N. et al. Clinical and echocardiographic disease
in patients starting end-stage renal disease therapy. Kidney Int
1995; 47: 186–92.
- London GM. Cardiovascular Disease in Chronic Renal Failure:
Pathophysiologic Aspects. Semin Dial 2003; 16(2): 85-94.
- Di Lullo L, Gorini A, Russo D, Santoboni A, Ronco C. Left
Ventricular Hypertrophy in Chronic Kidney Disease Patients: From
Pathophysiology to Treatment. Cardiorenal Med 2015; 5(4):
254–266.
- Testa A, Mallamaci F, Benedetto F, Pisano A, Tripepi G,
Malatino L, Thadhani R, Zoccali C. Vitamin D receptor (VDR) gene
polymorphism is associated with left ventricular (LV) mass and
predicts left ventricular hypertrophy (LVH) progression in
end-stage renal disease (ESRD) patients. J Bone Miner Res 2010;
25: 313–319.
- El-Shehaby AM, El-Khatib MM, Marzouk S, Battah AA.
Relationship of BsmI polymorphism of vitamin D receptor gene
with left ventricular hypertrophy and atherosclerosis in
hemodialysis patients. Scand J Clin Lab Invest 2013; 73: 75–81.
- Sárközy M, Gaspar R, Zvara A, Siska A, Kővári B, Szűcs G, et
al. Chronic kidney disease induces left ventricular
overexpression of the pro-hypertrophic microRNA-212. Scientific
Reports 2019; 9: 1302|
https://doi.org/10.1038/s41598-018-37690-5
- Green D, Roberts PR, New DI, Kalra PA. Sudden cardiac death
in hemodialysis patients: an in-depth review. Am J Kidney Dis
2011; 57: 921-9.
- Ketteler M, Schlieper G, Floege J. Calcification and
Cardiovascular Health. New Insights Into an Old Phenomenon.
Hypertension 2006; 47(6): 1027–34.
- Salusky IB, Goodman WG. Cardiovascular calcification in
end-stage renal disease. Nephrol Dial Transplant 2002; 17:
336-9.
- Giachelli, Cecilia M. The emerging role of phosphate in
vascular calcification. Kidney International 2009; 75 (9):
890-7.
- Azpiazua D, Gonzaloa S, González-Parrab E, Egidoa J,
Villa-Bellosta R. Role of pyrophosphate in vascular
calcification in chronic kidney disease. Nefrologia 2018; 38(3):
250-7.
- Amarpali Brar, Jeans M. Santana, Moro O. Salifu and Clinton
D. Brown (January 16th 2019). Dyslipidemia in Special
Populations, the Elderly, Women, HIV, Chronic Kidney Disease and
ESRD, and Minority Groups [Online First], IntechOpen, DOI:
10.5772/intechopen.82831.
- Kronenberg F. HDL in CKD: the devil is in the detail. J Am
Soc Nephrol 2018; 29:1356–71.
- Chang TI, Streja E, Soohoo M, Ko GJ, Rhee CM, Kovesdy CP, et
al. Increments in serum high-density lipoprotein cholesterol
over time are not associated with improved outcomes in incident
hemodialysis patients. J Clin Lipidol 2018;12(2):488-97.
- Dai L, Golembiewska E, Lindholm B, Stenvinkel P. End-Stage
Renal Disease, Inflammation and Cardiovascular Outcomes. Contrib
Nephrol 2017; 191: 32-43.
- Ronco C, Haapio M, House AA, Anavekar N, Bellomo R.
Cardiorenal syndrome. J Am Coll Cardiol 2008; 52: 1527-39.
- Van Laecke S, Van Biesen W. Smoking and chronic kidney
disease: seeing the signs through the smoke? Nephrol Dial
Transplant 2017; 32(3): 403–5.
- Xia J, Wang L, Ma Z, Zhong L, Wang Y, Gao Y, et al.
Cigarette smoking and chronic kidney disease in the general
population: a systematic review and meta-analysis of prospective
cohort studies. Nephrol Dial Transplant 2017; 32(3): 475-87.
- Nakamura K, Nakagawa H, Murakami Y, Kitamura A, Kiyama M,
Sakata K, et al. Smoking increases the risk of all-cause and
cardiovascular mortality in patients with chronic kidney
disease. Kidney Int 2015; 88: 1144–52.
- Roehm B, Simoni J, Pruszynski J, Wesson D.E. Cigarette
Smoking Attenuates Kidney Protection by Angiotensin-Converting
Enzyme Inhibition in Nondiabetic Chronic Kidney Disease. Am J
Nephrol 2017; 46: 260-7.
- Mallamaci F, Tripepi G. Salt restriction in chronic kidney
disease: a simple need or a must? Kidney Blood Press Res 2014;
39(2-3): 124-8.
- Kidney Disease: Improving Global Outcomes (KDIGO) CKD Work
Group. KDIGO 2012 clinical practice guideline for the evaluation
and management of chronic kidney disease. Kidney Int 2013; 3:
1–150
- Mente A, O’Donnell M, Rangarajan S, Dagenais G, Lear S,
McQueen M, et al. Associations of urinary sodium excretion with
cardiovascular events in individuals with and without
hypertension: a pooled analysis of data from four studies.
Lancet 2016; 388: 465–75
- Ko GJ, Obi Y, Tortorici AR, Kalantar-Zadeh K. Dietary
protein intake and chronic kidney disease. Curr Opin Clin Nutr
Metab Care 2017; 20(1): 77–85.
- Bellizzi V, Carrero JJ, Chauveau P, Cozzolino M, Cupisti A,
D'Alessandro C, et al. Retarding chronic kidney disease (CKD)
progression: a practical nutritional approach for non-dialysis
CKD, European Renal Nutrition Working Group of the European
Renal Association-European Dialysis Transplant Association
(ERA-EDTA). Nephrol Point Care 2016; 2: 56–67.
- Stenvinkel P, Zoccali C, Ikizler TA. Obesity in CKD--what
should nephrologists know? J Am Soc Nephrol 2013; 24(11):
1727–36.
- Postorino M, Marino C, Tripepi G, Zoccali C, for the CREDIT
(Calabria Registry of Dialysis and Transplantation) Working
Group. Abdominal obesity and all-cause and cardiovascular
mortality in end-stage renal disease. J Am Coll Cardiol 2009;
53: 1265-72.
- Navaneethan SD, Yehnert H, Moustarah F, Schreiber MJ,
Schauer PR, Beddhu S. Weight loss interventions in chronic
kidney disease: a systematic review and meta-analysis. Clin J Am
Soc Nephrol 2009; 4: 1565-74
- Johansen KL, Lee C. Body composition in chronic kidney
disease. Curr Opin Nephrol Hypertens 2015; 24: 268-75.
- WHO: Global Recommendations on Physical Activity for Health,
Geneva, World Health Organization, 2010;
- Robinson ES, Fisher ND, Forman JP, Curhan GC. Physical
activity and albuminuria. Am J Epidemiol 2010; 171: 515-21.
- Stack AG, Molony DA, Rives T, Tyson J, Murthy BVR.
Association of physical activity with mortality in the US
dialysis population. Am J Kidney Dis 2005; 45: 690-701.
- Sinha AD, Agarwal R. The complex relationship between CKD
and ambulatory blood pressure patterns. Adv Chronic Kidney Dis
2015; 22(2): 102–7.
- Pugh D, Gallacher PJ, Dhaun N. Management of Hypertension in
Chronic Kidney Disease. Drugs 2019; 79(4): 365-79.
- Whelton PK, Carey RM, Aronow WS, Casey DE, Jr, Collins KJ,
Dennison Himmelfarb C, et al. 2017
ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA guideline for
the prevention, detection, evaluation, and management of high
blood pressure in adults: a report of the American College of
Cardiology/American Heart Association Task Force on Clinical
Practice Guidelines. J Am Coll Cardiol 2018; 71: e127–e248.
- National Institute for Health and Care Excellence. Chronic
kidney disease in adults: assessment and management. London:
NICE; 2014.
- The UK Renal Association. Hypertension. 2018.
https://renal.org/information-resources/the-uk-eckd-guide/hypertension/.
Accessed 1 Nov 2018.
- Taler SJ, Agarwal R, Bakris GL, Flynn JT, Nilsson PM, Rahman
M, et al. KDOQI US commentary on the 2012 KDIGO clinical
practice guideline for management of blood pressure in CKD. Am J
Kidney Dis 2013; 62: 201–13.
- Williams B, Mancia G, Spiering W, Agabiti Rosei E, Azizi M,
Burnier M, et al. 2018 ESC/ESH Guidelines for the management of
arterial hypertension: the Task Force for the Management of
Arterial Hypertension of the European Society of Cardiology and
the European Society of Hypertension: the Task Force for the
Management of Arterial Hypertension of the European Society of
Cardiology and the European Society of Hypertension. J Hypertens
2018; 36: 1953–2041.
- Badve SV, Roberts MA, Hawley CM et al. Effects of
beta-adrenergic antagonists in patients with chronic kidney
disease: a systematic review and meta-analysis. J Am Coll
Cardiol 2011; 58: 1152–61.
- Cooper ME. Glucose lowering and kidney protection: can we
hit 2 birds with 1 stone? Medicographia 2013; 114 (35): 48-52.
- Boussageon R, Pouchain D, Renard V. Prevention of
complications in type 2 diabetes: is drug glucose control
evidence based? Br J Gen Pract 2017; 67(655): 85–7.
- Boussageon R, Bejan-Angoulvant T, Saadatian-Elahi M, et al.
Effect of intensive glucose lowering treatment on all cause
mortality, cardiovascular death, and microvascular events in
type 2 diabetes: meta-analysis of randomised controlled trials.
BMJ 2011; 343: d4169.
- Bongaerts B, Lindner LM, Hoyer A, Kuss O, Herder C,
Al-Hasani H. Mortality Risk of Noninsulin Glucose-Lowering Drugs
in Type 2 Diabetes—A Network Meta-analysis of Observational
Trials.Diabetes 2018; 67(Suppl1): 1139-P
- Maqbool M, Cooper ME, Jandeleit-Dahm KAM. Cardiovascular
Disease and Diabetic Kidney Disease. Semin Nephro. 2018;38(3):
217-32.
- Baigent C, Landray MJ, Reith C, et al, for the SHARP
Investigators. The effects of lowering LDL cholesterol with
simvastatin plus ezetimibe in patients with chronic kidney
disease (Study of Heart and Renal Protection): a randomised
placebo-controlled trial. Lancet 2011; 377: 2181–92.
- Palmer SC, Craig JC, Navaneethan SD, Tonelli M, Pellegrini
F, Strippoli GF. Benefits and harms of statin therapy for
persons with chronic kidney disease: a systematic review and
meta-analysis. Ann Intern Med 2012; 157: 263–75.
- Palmer SC, Di Micco L, Razavian M, Craig JC, Perkovic V,
Pellegrini F, et al. Antiplatelet agents for chronic kidney
disease. Cochrane Database Syst Rev 2013; (2):CD008834.
- Chan KE, Lazarus JM, Thadhani R, Hakim RM. Warfarin use
associates with increased risk for stroke in hemodialysis
patients with atrial fibrillation. J Am Soc Nephrol 2009; 20:
2223-33.
- U.S. Preventive Services Task Force. Aspirin for the
prevention of cardiovascular disease: U.S. Preventive Services
Task Force Recommendation Statement. Ann Intern Med. 2009; 150:
396–404
- Smith, S.C. Jr., Benjamin, E.J., Bonow, R.O. et al. AHA/ACCF
secondary prevention and risk reduction therapy for patients
with coronary and other atherosclerotic vascular disease: 2011
update: a guideline from the American Heart Association and
American College of Cardiology Foundation. Circulation. 2011;
124: 2458–73
- Lankhorsta CE, Wish JB. Anemia in renal disease: Diagnosis
and management. Blood rev 2010; 24(1): 39-47.
- Berns JS. Cardiovascular and renal effects of anemia in
chronic kidney disease.
https://www.uptodate.com/contents/cardiovascular-and-renal-effects-of-anemia-in-chronic-kidney-disease
- Locatelli F, Del Vecchio L, Pozzoni P. Anemia and
cardiovascular risk: the lesson of the CREATE Trial. J Am Soc
Nephrol. 2006;17(12 Suppl 3): S262-6.
- Singh AK, Szczech L, Tang KL, Barnhart H, Sapp S, Wolfson M,
et al; CHOIR Investigators. Correction of anemia with epoetin
alfa in chronic kidney disease. N Engl J Med. 2006; 355(20):
2085-98.
- Pfeffer MA, Burdmann EA, Chen CY, Cooper ME, de Zeeuw D,
Eckardt KU et al; TREAT Investigators. A trial of darbepoetin
alfa in type 2 diabetes and chronic kidney disease. N Engl J
Med. 2009; 361(21): 2019-32.
- Yuen NK, Ananthakrishnan S, Campbell MJ. Hyperparathyroidism
of Renal Disease. Perm J 2016; 20(3): 15–127.
- Cozzolino M, Mangano M, Stucchi A, Ciceri P, Conte F,
Galassi A. Cardiovascular disease in dialysis patients. Nephrol
Dial Transplant 2018; 33(suppl_3): iii28–iii34.
- Scialla JJ, Wolf M. Roles of phosphate and fibroblast growth
factor 23 in cardiovascular disease. Nat Rev Nephrol 2014;
10(5): 268-78.
- Isakova T, Gutierrez OM, Chang Y, et al. Phosphorus binders
and survival on hemodialysis. J Am Soc Nephrol 2009; 20(2):
388–96.
- Chang TI, Abdalla S, London GM, Block GA, Correa-Rotter R,
Drüeke TB, et al. The effects of cinacalcet on blood pressure,
mortality and cardiovascular endpoints in the EVOLVE trial. J
Hum Hypertens 2016; 30(3): 204-9.
- Kim CS, Jin DC, Yun YC, Bae EH, Ma SK, Kim SW. Relationship
between serum uric acid and mortality among hemodialysis
patients: Retrospective analysis of Korean end-stage renal
disease registry data. Kidney Res Clin Pract 2017; 36(4):
368–76.
- Raikou VD, Kyriaki D. Association between low serum
bicarbonate concentrations and cardiovascular disease in
patients in the end-stage of renal disease. Diseases 2016; 4: 36
- Voiculeț C, Zară O, Bogeanu C. et al. The role of oral
sodium bicarbonate supplementation in maintaining acid-base
balance and its influence on the cardiovascular system in
chronic hemodialysis patients - results of a prospective study.
J Med Life 2016; 9: 449–54
|
|
|
|
