|
||||||||||||||||||||||||||||||||||||
| [ Sadržaj
] [ Indeks autora ]
|
||||||||||||||||||||||||||||||||||||
| UDK 502/504:061.3(497.11)"1993/2017" COBISS.SR-ID 271719180 |
ISSN 0350-2899. - Vol. 43, br. 3 (2018), str. 132-145 | |||||||||||||||||||||||||||||||||||
|
Istorija medicine / History of medicine Razumevanje problema
fenilketonurije kroz istoriju
Biljana Stojanović Jovanović, Stevan Jovanović |
||||||||||||||||||||||||||||||||||||
|
|
||||||||||||||||||||||||||||||||||||
| Preuzmite rad u pdf formatu |
Sadržaj: Fenilketonurija (PKU) je autozomno-recesivna
metabolička bolest koja dovodi do mentalne retardacije i drugih
neuroloških problema kada se lečenje ne započne u roku od prvih
nekoliko nedelja života. Nastaje zbog nedostatka enzima
fenilalanin-hidroksilaze (PAH), pa se fenilalanin nagomilava u
organizmu, a smanjuje nivo tirozina. Prevelika količina fenilalanina
u krvotoku dovodi do oštećenja mozga kod dece. Nedovoljna količina
tirozina dovodi do smanjenja produkcije pigmenta melanina, tako da
su deca sa PKU plava, nežne bele kože, sa plavim očima. Fenilalanin
se u obliku fenilketona izlučuje urinom. Usled ovih ketona, znoj i
urin obolelih imaju jači miris nego zdrave osobe (podseća na buđ).
Gatrijev test se sprovodi kao masovna, skrining metoda za
fenilketonuriju. i on je obavezni nacionalni program zdravstvene
zaštite dece. Lečenje se sprovodi dijetom koja ograničava unos
fenilalanina. Summary: Phenylketonuria (PKU) is an autosomal recessive,
metabolic disease that leads to mental retardation and other
neurological problems, when treatment is not started within the
first few weeks of life. It occurs due to the lack of phenylalanine
hydroxylase enzyme (PAH) and phenylalanine is accumulated in the
body, and the tyrosine level decreases. An excessive amount of
phenylalanine in the bloodstream leads to brain damage in children.
Insufficient tyrosine leads to a decrease in the production of
melanin pigments, so children with PKU are blond, with soft white
skin and blue eyes. Phenylalanine is excreted in the form of
phenylketone in the urine. Due to these ketones, sweat and urine of
sufferers have a stronger smell than of a healthy persons
(resembling budding). The Guthrie test is carried out as a mass,
screening method for phenylketonuria and it is a part of mandatory
national child health care program. Treatment is carried out by a
diet that limits the intake of phenylalanine. |
|||||||||||||||||||||||||||||||||||
UVODFenilketonurija (PKU) je najčešća urođena bolest metabolizma
aminokiselina. Prenosi se autozomno recesivnim putem. Bolest nastaje
kao posledica nedostatka enzima fenilalanin-hidroksilaze u jetri.
Enzim fenilalanin-hidroksilaza metaboliše fenilalanin u tirozin.
Zbog nedostatka enzima fenilalanin- hidroksilaze, ako je enzim
defektan ili nepostojeći, ova reakcija ne može da se realizuje i
dolazi do nagomilavanja fenilalanina u organizmu, a smanjuje se nivo
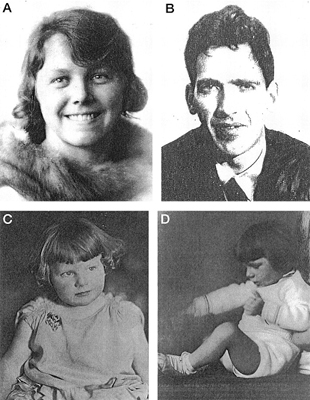
tirozina [1-6]. NAJRANIJI OPISU medicinsku literaturu pojam nasledne metaboličke bolesti, uveo je engleski lekar Archibald Edward Garrod 1908 godine [14]. On je uočio da je bolest urođena i češća kod bliskih srodnika pa je na osnovu toga zaključio da je nasledna. Svoja istraživanja objavio je u radu Urođene bolesti metabolizma (Inborn errors of metabolism.) koji je objavljen 1909. godine [15]. Fenilketonuriju je prvi put opisao norveški lekar i naučnik Ivar Asbjorn Foling 1934. godine (slika 1). Slika 1. Ivar Asbjorn Folling
Doktoru Folingu, koji je u tadašnje vreme bio na dobrom glasu kao poznavalac biohemije, obratila se jedna majka dvoje mentalno zaostale dece nakon dugotrajnog obilaženja i traženja pomoći jer je htela da sazna, da li je čudan miris urina njene dece povezan sa njihovom mentalnom zaostalošću. Devojčica je imala šest godina, znala je da izgovori samo nekoliko reči, imala je tipične znake spastične paralize sa uobičajenim teškoćama u kretanju. Mlađi brat od četiri godine nije govorio niti hodao, nije pratio pogledom predmete oko sebe i nije kontrolisao stolicu i mokrenje (slika 2). Slika 2. ,,Prva“ PKU porodica
Foling je analizirao njihov urin koristeći rastvor feri-hlorida,
očekujući da će dobiti crveno-braon boju karakterističnu za ketone.
Kod malih pacijenata čiji je urin doktor pregledao primetilo se da
je mokraća poprimila tamno zelenu boju za koju se zna da potiče od
fenilpiruvata, metabolita fenilalanina [16, 17]. Prvi zaključak je
bio da dvoje dece, sestra i brat sa umnom zaostalošću izlučuju u
mokraći nešto čega nema kod zdrave dece. Pitanja koja su se
nametnula: izlučuju li sva mentalno zaostala deca takvu mokraću i
koji je hemijski sastav mokraće te dece? Odgovor na ova pitanja
otkriva novu bolest, za koju je dr Foling predložio naziv
„fenilpiruvična idiotija“, time se želelo istaći da je reč o bolesti
praćenoj mentalnom retardacijom uz koju bolesnici izlučuju
fenilpiruvičnu kiselinu u mokraći [18]. Nekoliko godina kasnije,
Penrose i Quastel menjaju ime bolesti u „fenilketonurija“ čiji se
naziv zadržao do danas [19]. Dr Folling nažalost nije mogao pomoći
oboleloj deci, ali je svojim otkrićem učinio prvi veliki korak, jer
je povezao hemijske promene u mokraći obolele dece sa naslednim
oblikom umne zaostalosti [18]. On je nakon detaljne hemijske analize
urina mentalno retardiranih brata i sestre, zatražio od svih lekara
u blizini Osla da detaljno analiziraju urin pacijenata koji imaju
bilo kakav oblik mentalne retardacije. Ista analiza u mokraći je
urađena kod 430 osoba sa mentalnom retardacijom do tada nepoznatog
uzroka i kod osam osoba dobijen je isti rezultat kao kod dvoje
opisane dece. Dr Folling je primetio da ovi bolesnici pored mentalne
retardacije imaju i druge sličnosti a to su; prisustvo ekcema, niski
rast, široka ramena i spastičan hod. Ispitivanjem porodičnog stabla
došao je do zaključka da je u pitanju autozomno recesivno
nasleđivanje [3]. POČECI SKRININGA NA FENILKETONURIJUŠezdesetih godina prošlog veka, pojava programa skrininga
novorođenčadi predstavljala je pravu malu revoluciju u preventivnoj
medicini. Danas su takvi programi širom sveta opšte prihvaćeni. Da
bi se neka bolest uvrstila u program sistemskog traganja, a da za to
postoji i javno zdravstveno opravdanje potrebni su odredjeni
uslovi[20]. Slika 3. Robert Gatri
Ovo otkriće je otvorilo put ranom, neonatalnom otkrivanju bolesti. Skrining na fenilketonuriju u SAD počinje 1962. godine [23]. Kod nas skrining počinje u Dubrovniku 1966. godine, kada je organizovan na inicijativu Ministarstva zdravlja SAD-a, nakon što je Američka vlada odobrila novac za izvođenje 100 000 skrining testova u nekoliko većih porodilišta tadašnje Jugoslavije [24]. Program je vođen iz Instituta za zdravstven zaštitu majke i deteta Republike Srbije „Dr Vukan Čupić“ u Beogradu i bilo je otkriveno i uspešno lečeno desetoro dece sa ovim problemom. Tačni zapisi o rezultatima nisu dostupni, a nakon što su sredstva potrošena, prestao je sistematski rad u ranom otkrivanju fenilketonurije kod novorođenčadi. Važno je bilo da se šira javnost upoznala sa problemom fenilketonurije. Pilot program na teritoriji tadašnje Jugoslavije sproveli su Vulović i saradnici 1967. godine, dok organizovani skrining na ovu bolest u Srbiji počinje da se sprovodi od 1983. godine [25]. NEONATALNI SKRINING NA FENILKETONURIJU U POSLEDNJOJ DEKADI XX VEKANovorođenački skrining je postupak u okviru preventivne medicine
kojem je svrha rano otkrivanje bolesne novorođenčadi kod koje će
pravovremena dijagnoza i lečenje dovesti do značajnog smanjenja
smrtnosti, morbiditeta i invalidnosti. Uzorak krvi treba uzeti
svakom novorođenčetu. Slika 4. Kartica za neonatalni skrining
Na agaroznu ploču se stavlja mali krug filter papira sa osušenom kapi krvi i ukoliko je povećana koncentracija fenilalanina, fenilpiruvata i fenilacetata doći će do rasta bakterija na agaroznoj ploči. Metoda je jednostavna, rezultati su dostupni za 24 sata, ima malu grešku merenja i jeftina je (cena jednog Guthrie testa je oko 2 evra) [9, 30]. SPROVOĐENJE SKRININGA NA FENILKETONURIJU DANASSkriningom su obuhvaćena sva novorođena deca. Uzoroci krvi za
skrining uzimaju se na skrining karticama u porodilištu i/ ili na
neonatološkim odeljenjima gde je dete rođeno ili u bolničkim
ustanovama ukoliko je novorođenče zbog bolesti bilo hospitalizovano.
Za novorođenčad u intenzivnoj i poluintenzivnoj nezi skrining se
odlaže do 10. dana života i uzima se još jedan uzorak pre otpusta.
Kod prevremeno rođene dece prvi uzorci za skrining uzimaju se
najkasnije do 10- og dana života, a potom se uzimanje uzoraka
ponavlja u drugoj nedelji života. Ako novorođenče ne toleriše
ishranu, sa prvim uzorkom se šalje i napomena da novorođenče nije na
oralnoj ishrani. Drugi uzorak se uzima kada novorođenče postigne
enteralni unos u trajanju od 48 sati. Ukoliko novorođenče mora da
dobije transfuziju krvi, prvi uzorak se uzima pre transfuzije, a
drugi u uzrastu od sedam dana ili pre otpusta [31, 32]. FENILKETONURIJA SE LEČI DIJETOMOsnova lečenja klasične fenilketonurije je sprovođenje striktne,
niskoproteinske dijete, sa smanjenim, kontrolisanim unosom
fenilalanina. Da bi se sprečile teške posledice, dijetu je neophodno
početi što ranije, u prvim nedeljama života deteta [31, 32]. Stavovi
o neophodnom trajanju ovakvog načina ishrane tokom vremena su se
menjali. Dijeta je najstroža u prvim godinama života kada je i
razvoj deteta najintenzivniji. Ranije je smatrano da je dovoljno
dijetu sprovoditi do šeste godine života. Postepeno je ta granica
pomerana ka starijem uzrastu. ZAKLJUČAKDeca obolela od fenilketonurije (PKU) pre uvođenja neonatalnog skrininga bila su lišena svih mogućnosti za vođenje normalnog života. Lečenje treba započeti što pre, čim se bolest otkrije a nažalost ono traje doživotno. Uz pravovremeno započeto lečenje (u prvim nedeljama života) i redovno, tačno i neprekidno sprovođenje lečenja i kontrole, dete ima odlične izglede da se razvija i raste, te se ni po čemu neće razlikovati od svojih vršnjaka. Svaki prekid lečenja i zanemarivanje redovne kontrole neminovno dovodi do porasta fenilalanina u krvi sa svim posledicama. Unapređenje dijagnostike, nege i lečenja kao i sveobuhvatnog kvaliteta života obolelih od fenilketonurije zahteva angažovanje svih činilaca zdravstvenog sistema Republike Srbije. Za uspešno lečenje fenilketonurije zaslužni su zdravstveni profesionalci koji postavljaju dijagnozu kroz neonatalne skrining programe, pedijatri, medicinske sestre dijetetičari i osobe koje nadgledaju tretman lečenja fenilketonurije. LITERATURA:
|
||||||||||||||||||||||||||||||||||||
|
|
||||||||||||||||||||||||||||||||||||
| Adresa autora / Corresponding
address: Biljana Stojanović - Jovanović, isoka zdravstvena škola strukovnih studija u Beogradu, Cara Dušana 254, Beograd E-mail: biljananstojanovic@gmail.com |
Rad primljen: 6.7.2018. Elektronska verzija objavljena: 24.12.2018. |
|||||||||||||||||||||||||||||||||||
| [ Sadržaj
] [ Indeks autora ]
|
||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||