|
||||||||||||||||||||||||||||||||||||
| [ Sadržaj
] [ Indeks autora ]
|
||||||||||||||||||||||||||||||||||||
| UDK 616-056.76-076 |
ISSN 0350-2899, 36(2011) br.3 p.162-65 |
|||||||||||||||||||||||||||||||||||
|
Prikaz slučaja Uloga elektronske mikroskopije u dijagnostici Alportovog sindroma - prikaz slučaja (The role of electron microscopy in the diagnosis of Alport syndroma - Case report) Milena Potić Floranović (1) , Jelena Rajković (1), Miloš Kostić (1), Miloš Bogoslović (2), Branka Mitić (3), Vidojko Đorđević (3), Vojin Savić (1) (1) Institut za biomedicinska istraživanja Medicinskog fakulteta u nišu, (2) Dom zdravlja Doljevac, (3) Institut za nefrologiju i hemodijalizu Kliničkog centra u Nišu |
||||||||||||||||||||||||||||||||||||
|
|
||||||||||||||||||||||||||||||||||||
| Sažetak: Alportov sindrom (AS) predstavlja hereditarnu hroničnu bolest koja se karakteriše progresivnim hematuričnim nefritisom, oštećenjem sluha i promenama na očima. Ovu genetski heterogenu bolest karakteriše mutacija gena odgovornih za singtezu kolagena tipa IV, koji ulazi u sastav bazalnih membrana širom organizma. Najčešća je X-vezana dominantna forma, kod koje su simptomi blaži kod žena koje su heterozigotni nosioci nego kod muškaraca. Verovatnoća razvijanja terminalne bubrežne insuficijencije pre 40 godine je 80% kod dečaka i muškaraca. Pacijenti sa AS predstavljaju dobre kandidate za renalnu transplantaciju. Pacijentkinja je ispitivana kao potencijalni donor organa za svog sina, kome je postavljena dijagnoza AS. Glavni klinički simptom majke je oštećenje sluha, ali su biohemijski parametri bili normalni, izuzev mikrohematurije. Tkivni uzorak dobijen je perkutanom iglenom biopsijom. Svetlosnomikroskopski nalaz bio je bez specifičnih promena. Imunofluorescentni nalaz je bio u potpunosti negativan. Elektronskom mikroskopijom uočeno je fokalno stanjenje i zadebljanje glomerularne bazalne membrane, diskontinuitet i retikulacija laminae dense i dilatacija i retikulacija laminae rare navedene promene odgovaraju dijagnozi AS. AS kao i ostale hereditarne nefropatije ne mogu biti adekvatno dijagnostikovane bez upotrebe elektronske mi-kroskopije. Njome se mogu detektovati ultrastrukturne promene bazalne membrane glomerula, promene u me-zangijumu, Bowmanovoj kapsuli prstastim nastavcima podocita i drugim strukturama. AS predstavlja vodeći uzrok hereditarne bubrežne insuficijencije. Transplantacija sa živog rođaka, ako je donor heterozigotni nosilac može nositi rizik kako za primaoca tako i za recipijenta. Iako žene imaju blaže simptome, rizik za razvoj terminalne bubrežne insuficijencije se povećava sa starenjem. Ovo treba uzeti u obzir, budući da se majke pacijenata sa AS često dobrovoljno javljaju kao donori organa. Elektronska mikroskopija bubrežnog tkiva može omogućiti skrining potencijalnih donora mođu članovima porodice sa inače normalnom renalnom funkcijom. Pri proceni potencijalnih donora ultramikroskopske promene u bubrežnom parenhimu mogu se smatrati kriterijumom za isključivanje. Ključne reči: Alportov sindrom, elektronska mikroskopija, transplantacija bunbrega Napomena: sažetak na engleskom jeziku Note: Summary in English |
||||||||||||||||||||||||||||||||||||
|
|
||||||||||||||||||||||||||||||||||||
UVODAlportov sindrom (AS) predstavlja hereditarnu hroničnu bolest
koja se karakteriše progresivnim hematuričnim nefritisom, oštećenjem
sluha i promenama na očima. Ovu genetski heterogenu bolest
karakteriše mutacija gena odgovornih za sintezu kolagena tipa IV
koji ulazi u sastav bazalnih membrana širom organizma. Oštećenje
glomerularne bazalne membrane bubrega dovodi do hereditarnog
nefritisa, koji se karakteriše mikrohematurijom, proteinurijom i
progresijom ka hroničnoj bubrežnoj insuficijenciji. Oštećenje
bazalne membrane kohlee i kortijevog organa dovodi do
senzorineuralnog gubitka sluha. Oštećenje kapsule sočiva dovodi do
nastanka mrljaste retinopatije i deformacije sočiva sa posledičnim
oštećenjem vida. [1] Postoji 6 gena sa kojih se prepisuje
informacija za sintezu izotipova kolagena IV i ovi geni su semešteni
u parovima na tri odvojena hromozoma: hromozomu 13 (gen COL4A1 i
COL4A2), hromozmu 2 (gen COL4A3 i COL4A4) i X-hromozomu (COL4A5 i
COL4A6).[2] Najčešća je X-vezana dominantna forma koja se sreće u
85% slučajeva i kod koje su simptomi blaži kod žena koje su
heterozigotni nosioci nego kod muškaraca. Kod muškaraca sa AS rizik
za razvoj terminalne bubrežne insuficijencije do tridesete godine
iznosi 70% a do četrdesete 90%. Pacijenti sa AS predstavljaju dobre
kandidate za renalnu transplantaciju. [3,4,5] PRIKAZ SLUČAJAPacijentkinja starosti 57 godina ispitivana je kao potencijalni
donor organa za svog sina, kome je postavljena dijagnoza AS. Majka
je kao glavni klinički simptom imala oštećenje sluha koje datira iz
detinjstva. Biohemijski parametri (urea, kreatinin, opšti pregled
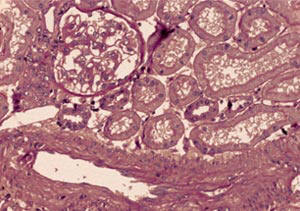
urina) bili su normalni, izuzev mikrohematurije. Tkivni uzorak
dobijen je perkutanom slepom iglenom biopsijom. Parafinski tkivni
kalupi su sečeni na debljinu od 4 mikrona i bojeni metodama:
hematoksilin eozin, PAS trihrom i Jones. Dobijeno tkivo je pripadalo
jukstamedularnom regionu kore bubrega a na parafinskim isečcima je
bilo prisutno ukupno 20 glomerula.
DISKUSIJAPrilikom ispitivanja renalnih biopsija elektronska mikroskopija potvrđuje i upotpunjuje dijagnozu postavljenu svetlosnomikroskopijom i imunofluorescencijom. Takođe može pružiti dodatne informacije od značaja za diferencijalnu dijagnozu. Međutim, AS i druge hereditarne nefropatije (sindrom tanke bazalne membrane s. benigna familijarna hematurija, fabrijeva bolest, nailpatella syndrome) ne mogu biti adekvatno dijagnostikovane bez upotrebe elektronskog mikroskopa [6,7].
U ovim slučajevima svetlosna mikroskopija i imunofluorescencija
ne mogu pružiti dovoljno informacija o patološkom procesu. I u
slučaju naše pacijentkinje klasične dijagnostičke metode (svetlosna
mikroskopija i imunofluorescenca) nisu pružile adekvatan uvid u
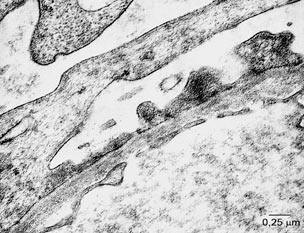
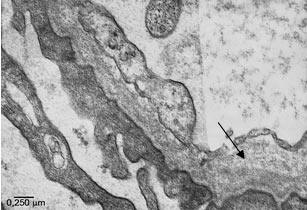
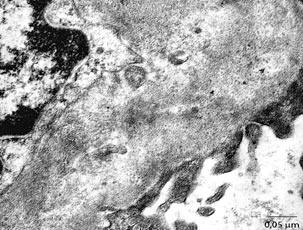
stanje glomerularne bazalne membrane. Elektronomikroskopski moguće
je sagledati i precizno lokalizovati ultrastrukturne promene
bubrežnog parenhima kao što su anoma-lije glomerularne bazalne
membrane, glomerularnih kapilara, mezangijuma, prisustvo imunih,
hijalinih ili fibrinskih depozita, oštećenje stopalastih nastavaka
podocita, Bowmanove kapsule i drugih komponenti. Merenje debljine
bazalne membrane glomerula može se vršiti jedino elektronskom
mikroskopijom. Smatra se da je u oko 18% urađenih biopsija neophodna
elektronska mikroskopija da bi se dijagnoza uopšte postavila, u oko
54% ona bitno doprinosi u donošenju konačne odluke patologa, a da u
28% slučajeva ovaj metod ispitivanja uopšte nema nikakvog značaja
[8,9]. Za dijagnozu AS pored elektronske mikroskopije pomoć može
pružiti i imunohistohemija, koja daje uvid o tipu kolagena koji
nedostaje. ZAKLJUČAKElektronska mikroskopija je tehnika čija je upotreba korisna u nefrologiji. U idealnom slučaju sve renalne biopsije kao i biopsije alograftova trebalo bi sagledati elektronskom mikroskopijom. Ona je neophodna za adekvatno postavljanje dijagnoze hereditarnih nefropatija. U transplantacionoj medicini elektronska mikroskopija može pružiti dodatne informacije osnovnom histopatološkom nalazu. LITERATURA
Rad je osvojio DRUGU NAGRADU Naučnog odbora XXX
Timočkih medicinskih dana |
||||||||||||||||||||||||||||||||||||
| Adresa autora: Milena Potić Floranović Medicinski fakultet, Niš Institut za biomedicinska istraživanja |
Rad primljen: 28.03.2011. Rad prihvaćen: 12.04.2011. Elektronska verzija objavljena: 17.12.2011. |
|||||||||||||||||||||||||||||||||||
| [ Sadržaj
] [ Indeks autora ]
|
||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||